 2018, Vol. 39
2018, Vol. 39



2. 浙江海洋高效健康养殖协同创新中心, 宁波 315211
2. Collaborative Innovation Center for Zhejiang Marine High-Efficiency and Healthy Aquaculture, Ningbo 315211, China
沿海城市工、农业和水产养殖业日益增加的排放, 导致沿海环境受到越来越严重的有机污染和营养盐的威胁, 给生态环境带来严重危害[1, 2].有机污染物能促进病原微生物的繁殖, 导致养殖环境生态系统失衡, 威胁沿海渔业发展.此外, 有机污染物导致水华的发生, 藻类分泌的毒素能够对浮游动物和底栖生物造成毒害作用.微生物起着原位生物传感器的作用, 能够用于评估自然或人为活动对环境的影响[3].因此, 微生物对污染状况的响应一直是研究者关注的焦点.
微生物在全球生物地球化学循环中起关键作用, 为其它生产者和消费者提供了营养[4].同时, 微生物群落通常是环境扰动的首要响应者, 能快速响应环境压力[5], 是最为敏感并极易受环境影响的生物类群[6].即使污染物本身已经完全降解, 微生物指示的应用也可系统地反映和记录生物和非生物(包括未测量和不可观察因素)的压力[7].例如, 在近海水产养殖中, 微生物群落结构和多样性有可能作为评价海水养殖生态系统健康和稳定的重要指标[8].此外, 有研究表明在养殖排放的有机污染物条件下, 潜在条件致病菌黄杆菌(Flavobacteriaceae)在养殖环境中大量存在, 可能导致虾病的暴发[9], 弧菌(Vibrionaceae)、交替假单胞菌(Pseudoalteromonas)等致病菌, 可以用作指示菌预测水产养殖病害状况[10].因此, 研究浮游细菌群落对污染的响应既是重要的环境问题又是重要的水产养殖问题.
浙江北部沿海水域毗邻中国最发达的两个区域(浙江和上海), 是长三角经济区的主体.杭州湾海域是典型的海陆过渡带, 自然变化剧烈, 人为干扰严重.随着人口的快速增长和海湾工业的集约发展, 渔业、海洋运输业以及勘探开采等带来的污染, 造成杭州湾水生生态系统健康严重恶化[11].大量陆地生物投入, 增加水体营养成分的同时, 降低了盐碱度.此外, 关于长江口及其邻近海域富营养化、生物多样性、浮游植物、重金属分布等报道相对较多[12, 13].有研究表明人类活动和陆源输入是影响浮游细菌分布的决定性因素[14], 而针对有机污染对浮游细菌群落的影响还有待进一步研究.因此, 本文通过Illumina MiSeq测序技术分析杭州湾及其邻近外海共21个站点不同有机污染程度对浮游细菌群落的影响, 利用有机污染指示值(A)评价海域污染状况.以期回答以下问题:①揭示不同有机污染程度对浮游细菌群落组成和多样性的影响; ②探讨浮游细菌群落与环境因子间的关系; ③能否筛选出敏感浮游细菌指示海域有机污染程度.
1 材料与方法 1.1 实验设计与采样杭州湾海域位于浙江东北部, 每年有大量的有机污染物输入, 造成沿径流的污染物梯度.为研究有机污染物对浮游细菌群落的影响, 本文选取了21个站点(30.37°~30.83°N, 121.21°~122.65°E), 其中包括杭州湾13个站点(H10、H11、H12、H20、H21、H24、H25、H27、H28、H31、H33、H36、H42)及其邻近外海8个站点(Z3、Z7、Z8、Z9、Z14、Z22、Z23、Z30).样品的采集、贮存、运输和预处理均按《海洋调查规范》(GB/T 12763-2007)和《海洋监测规范》(GB 17378-2007)中规定的标准方法进行.
1.2 海水理化指标测定现场测定海水温度、pH、溶解氧(DO)和盐度, 其它指标均按《海洋监测规范》(GB 17378.4-2007)所规定的标准方法进行:化学需氧量(COD)用碱性高锰酸钾法、悬浮颗粒(SP)用重量法、硝酸盐(NO3-)用锌-镉还原法、磷酸盐(PO43-)用抗坏血酸还原磷钼蓝法、氨盐(NH4+)用次溴酸钠氧化法、亚硝酸盐(NO2-)用重氮-偶氮法、油类用紫外分光光度法、总有机碳(TOC)用总有机碳仪器法、总氮(TN)和总磷(TP)用过硫酸钾氧化法、硅酸盐(SiO3-)用硅钼蓝法、砷和汞用原子荧光法、铜铅镉锌都用火焰原子吸收分光光度法连续测定、用二苯碳酰二肼分光光度法测定铬, 叶绿素a(Chla)用丙酮萃取分光光度法.
1.3 DNA提取, 细菌16S扩增和Illumine MiSeq测序水样经100 μm孔径的尼龙网预过滤, 然后用0.22 μm孔径的聚碳酸酯膜(Millipore, Boston, MA, USA)负压抽滤收集微生物.滤膜用无菌剪刀剪碎后用Power Soil ® DNA试剂盒(MO BIO Laboratories, Carlsbad, CA, USA)提取总DNA. NanoDrop ND-1000分光光度计测定DNA的浓度和纯度, 抽提后的DNA于-80℃保存备用.
每个样品取100 ng的DNA作为模板, 用F515(5′-GTGCCAGCMGCCGCGGTAA-3′)和R806(5′-GGACTACHVGGGTWTCTAAT-3′)引物扩增细菌16S rRNA基因的V4区, 正向引物5′端带有6 bp的Barcode序列.每个样品设置3个PCR重复, 以降低扩增造成的偏差. PCR的反应条件如下:94℃变性3 min, 34个循环, 94℃变性30 s, 55℃退火30 s, 72℃延伸30 s, 最后在72℃延伸8 min.扩增产物经1%琼脂糖凝胶电泳检测, 得到与扩增预期大小一致且没有非特异性条带后, 用PCR片段纯化试剂盒(Takara Biotech, Japan)进行纯化.通过分光光度计测定浓度后, 每个样品取等量的PCR产物混合, 并用MiSeq双末端测序平台测序(Illumina, San Diego, USA).
1.4 Illumina数据处理下机测序数据用FLASH软件拼接二端序列[15], 利用Qiime(quantitative insights into microbial ecology, http://qiime.org/)流程分析, 包括去嵌合体和质控[16].简言之, 剪接掉3个以上的连续且质控分小于20的碱基, 保留>300 bp的长序列, 然后用UCHIME去掉嵌合体.用UCLUST在97%的相似性水平聚类为分类操作单元(operational taxonomic unit, OTUs)[17].选取每个OTU中丰度和覆盖度最高的序列作为代表序列, 用PyNAST在Greengenes 13.8数据库中比对, 获得OTUs的物种分类信息[18, 19], 去除古菌、叶绿体、不属于细菌和在所有样品中只出现一次的序列.为避免测序深度不同造成多样性估计的偏差, 每个样品随机选取10 000条序列(最低测序深度)进行后续分析.
1.5 有机污染采用有机污染综合指数法(A)对该海域的污染状况进行评估, 有机污染指数计算公式为:
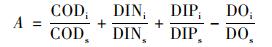
|
式中, A为有机污染指数, CODi、DINi、DIPi、DOi分别为化学需氧量、无机氮(包括铵态氮、硝态氮和亚硝态氮)、活性磷酸盐和溶解氧的实测值, CODs、DINs、DIPs、DOs分别为相应要素的一类海水其值依次为2.0、0.2、0.015和6.0 mg·L-1[20].
1.6 统计分析用SPSS 13.0做独立样本t检验比较杭州湾及其邻近外海环境参数、浮游细菌多样性的差异显著性.基于Bray-Curtis距离通过非度量多维尺度分析(non-metric multidimensional scaling, NMDS)不同站位点浮游细菌群落间的差异; 一般采用压力系数来评定所得到的多维构型与实际数据之间的适合度, 压力系数越小, 表示适合度越高[21].多元回归树筛选影响细菌多样性的环境因子[22].利用BioEnv分析筛选出对浮游细菌群落变异最相关的环境因子组合, 并用冗余分析表征环境因子与细菌群落变异的相关性[23].本研究主要关注有机污染对浮游细菌组成和多样性的影响, 利用R软件中的“labdsv”软件包筛选指示不同有机污染程度的敏感浮游细菌群落, 进一步通过Pearson相关性分析挑选出的指示群与有机污染间的差异显著性, 并用热图(Heatmap)显示在各站点中的分布.
2 结果与分析 2.1 杭州湾及其邻近外海海水环境参数检测的海水环境参数显示营养盐(P < 0.05, 除氨氮外)分布从近岸向外海逐渐递减, 有机污染A、悬浮颗粒、盐度、总有机碳、油类均有显著差异(P < 0.05, 表 1), 且杭州湾海域的悬浮颗粒高于其邻近外海约10倍, 使得其成为最大的浊度带.
|
|
表 1 独立样本t检验分析杭州海域及其邻近外海环境参数的差异1) Table 1 Independent sample t test analysis of the difference in environmental parameters in Hangzhou Bay and the adjacent sea areas |
2.2 浮游细菌群落组成和结构分析
杭州湾海域及其邻近外海水体中主要细菌类群为:α-变形菌纲(α-Proteobacteria)、β-变形菌纲(β-Proteobacteria)、γ-变形菌纲(γ-Proteobacteria)、δ-变形菌纲(δ-Proteobacteria)、浮霉菌门(Planctomycetes)、放线菌门(Actinobacteria)、蓝细菌门(Cyanobacteria)、拟杆菌门(Bacteroidetes), 对应杭州湾平均相对丰度分别为16.5%、5.8%、24.4%、4.9%、13.9%、9.4%、1.1%、6.9%, 合计占总细菌序列数的82.9%;邻近外海平均相对丰度分别为14.3%、1.7%、16.6%、1.4%、4.1%、17.5%、20.1%、18.4%, 合计占总细菌序列数的94.1%(图 1).杭州湾的浮霉菌门、β-变形菌纲显著(P < 0.001, t检验, 下同)高于邻近外海, 而放线菌门(P=0.012)、蓝细菌门(P=0.002)和拟杆菌门(P=0.017)的相对丰度则显著较低.

|
图 1 杭州湾及其邻近外海主要优势菌群平均相对丰度 Fig. 1 Relative abundances of the dominant communities in Hangzhou Bay and its adjacent offshore sea |
基于样品中检测到的OTUs丰度, NMDS分析表明浮游细菌群落按站位点聚类, 杭州湾水域与对照区浮游细菌群落组成有明显差异[图 2(a)].同时群落相似性检验表明两区域浮游细菌组成差异显著(r=0.916, P < 0.001).此外, 回归拟合分析表明细菌群落距离与有机污染程度显著相关(P < 0.001)[图 2(b)], 表明细菌群落的变化取决于有机污染所产生的选择压力.

|
图 2 非度量多维尺度分析浮游细菌群落组成以及群落距离与有机污染的对数拟合 Fig. 2 Nonmetric multidimensional scaling analysis of bacterioplankton community composition and logarithm regression of community distance and organic pollution |
杭州湾海域浮游细菌群落的多样性, 包括物种多样性(observed species)、香农多样性(Shannon diversity)和系统发育多样性(phylogenetic diversity)均显著(P < 0.001)高于邻近外海对照区(表 2).浮游细菌多样性差异主要与悬浮颗粒、硝酸盐、磷酸盐、总磷和有机污染有关, 其中与有机污染和悬浮颗粒的相关性最高(表 3).
|
|
表 2 浮游细菌群落多样性指数 Table 2 Diversity indices of bacterioplankton community |
|
|
表 3 浮游细菌多样性或群落组成与环境因子间的相关性分析1) Table 3 Correlation between bacterioplankton diversity or composition and environmental factors |
利用多元回归树分析发现悬浮颗粒、油污、亚硝酸盐、叶绿素a是影响浮游细菌群落多样性的主要因素(分别解释了22.0%、6.0%、6.5%、4.5%的多样性变异), 其次为pH、有机污染A和化学需氧量(分别解释了4.1%、5.5%、4.8%的多样性变异)(图 3).

|
每片叶子(终端)数值分别为相对误差和所含样品的数量 图 3 多元回归树分析影响浮游细菌多样性的环境因子 Fig. 3 Multiple regression tree analysis for identification of factors shaping the bacterioplankton diversity |
对环境因子进行标准化处理, Mantel检验(Mantel test, permutation=999)分析表明杭州湾海域及邻近外海微生物群落组成与有机污染最相关(r=-0.942, P < 0.001)(表 3).采用BioEnv筛选出对群落变异最相关(r=0.82)的环境因子组合为有机污染A、化学需氧量、叶绿素a、总氮、硝酸盐和盐度.冗余分析第一排序轴(RDA 1)的主要环境因子为有机污染A(负相关, r=-0.95)、硝酸盐(负相关, r=-0.92)、总氮(负相关, r=-0.93)、叶绿素a(正相关, r=0.70);第二排序轴(RDA 2)的主要环境因子为盐度(正相关, r=0.86)、化学需氧量(负相关, r=-0.61), 这些因子共同解释了71%的群落变异.有机污染作为单因子控制了6.5%, 浮游细菌群落变异较大(图 4).

|
图 4 浮游细菌群落及其变异驱动的主要环境因子RDA双向排序图 Fig. 4 Redundancy analysis (RDA) ordination biplot showing the factors that drive the variations in bacterial community |
采用指示值法共挑选出35个敏感物种(在OTUs水平), 利用热图显示指示物种的分布特征, 结果显示各样本能够按站点进行聚类(图 5), 区分效果与NMDS分析相类似[图 2(a)].杭州湾海域主要指示物种为立克次氏体(Rickettsiales)、浮霉菌门、硝化螺旋菌科(Nitrospiraceae)、酸杆菌门(Acidobacteria)、甲基球菌科(Methylococcaceae)和黄单胞杆菌目(Xanthomonadales), 而OM60、红杆菌科(Rhodobacteraceae)、黄杆菌科、蟑螂杆状体科(Cryomorphaceae)和放线菌门在其邻近外海相对丰度较高.此外, Pearson相关性分析表明指示群相对丰度与有机污染显著相关(P < 0.05)(图 5).因此, 这些细菌为评价海域有机污染水平提供了敏感的生物学指标.

|
图 5 与有机污染显著相关的35个指示物种 Fig. 5 Indicator species significantly associated with the organic pollution index |
有机污染物严重的杭州湾海域与邻近外海对照区的浮游细菌群落组成差异显著, 有机污染程度随群落距离的增加而减少(图 2), 但细菌多样性随有机污染的增加而增加.其主要优势菌群为α-变形菌纲、γ-变形菌纲、蓝细菌和拟杆菌(图 1).其它研究也发现拟杆菌门和变形菌门等异养细菌在海洋中占较高比例[24, 25].随着有机污染程度增加, α-变形菌纲、γ-变形菌纲和浮霉菌门的相对丰度也随之上升, 而拟杆菌和蓝细菌恰好相反.拟杆菌能够降解复杂有机物, 是有机物质的主要降解者[26], 这与其在寡营养环境的邻近外海相对丰度较高相一致(图 1).因此, α-变形菌纲、γ-变形菌纲、蓝细菌和拟杆菌的相对丰度对水质变化敏感, 可能对于指示和评估海域养殖有机污染引起的生态效应有一定的作用.多元回归树分析表明悬浮颗粒解释了22%的多样性变异, 是最主要的驱动因子(图 3), 这与悬浮颗粒与多样性指数强相关性结果相一致(表 3).有研究表明, 悬浮颗粒在不同的微生境存在不同的可用基质, 而更高的多样性可能与微生物相关的生态位丰富度增加有关, 解释了本研究中浮游细菌多样性与悬浮颗粒的最强耦合[27].
3.2 影响浮游细菌群落的主要环境因子杭州湾地处径流带及主要工业区, 工业废水和生活污水大量入海, 再加上两河流域大量水和泥沙排放, 水体中的悬浮颗粒物及死亡的动植物及代谢产物等通过一系列降解矿化成无机盐, 同时也造成了一定的石油类和金属污染, 其邻近水域有机污染严重程度不断增加.本检测结果也发现营养盐(除氨氮)、悬浮颗粒、油等在杭州湾海域显著(P < 0.05)高于邻近外海对照区(表 1).主要营养盐分布从近岸向外海逐渐递减, 而该海域大量营养盐的输入, 使浮游植物种间相互作用增加, 导致群落结构改变, 优势种形成[11].丰富的营养盐也会使藻类过度生长, 与本研究在邻近外海检测到的较高叶绿素a浓度结果一致.在中、低盐度区域, 如杭州湾, 高浓度的悬浮颗粒对磷酸盐表现出吸附-解吸的“缓冲”现象[28].悬浮物对海域叶绿素a的分布有直接影响, 悬浮物含量低的海域叶绿素a浓度相对较高[11].而邻近海域叶绿素a含量高也可能是由于蓝细菌相对丰度较高(图 1).
利用BioEnv分析表明有机污染物、化学需氧量、叶绿素a、总氮、硝酸盐和盐度是影响浮游细菌群落的主要环境因子组合.有研究发现细菌群落的分布与有机污染程度相关[29], 这与本研究的结果相一致, 且相比于其他环境因子, 有机污染作为单一因子解释了6.5%的群落变异, 对浮游细菌群落变化影响较大(图 4).化学需氧量是衡量水中有机物质含量多少的一个综合指标, 水体中的溶解性有机物是异养细菌生长繁殖和生命活动最基本的营养源[30].而近海高密度海水养殖, 往往忽视了例如未被利用的饵料、养殖水生生物自身的代谢产物等会产生大量有机质, 造成化学需氧量不同而影响浮游细菌群落.类似地, 营养盐浓度随排放距离逐渐降低, 其中硝酸盐和硅酸盐变化差异显著(表 1).有大量的研究证实, 营养盐浓度的动态变化会使某些浮游植物成为优势种, 种间竞争压力增加[11].此外, 高密度水产养殖也会引起海水环境中N、P等营养元素的增加, 影响浮游细菌群落组成, 促进了条件致病菌的增殖和水产养殖病害的发生.在全球范围内, 盐度已被认为是海洋环境中浮游细菌群落组成的主要决定因素[31]; 大量研究报道了在不同河口盐度降低会导致β-变形杆菌数量的增加[32, 33]; 在沿海环境中盐度可能有助于物理分离水体及其驻留细菌群落的密度梯度[34].
3.3 与有机污染相关的敏感指示物种微生物能够综合地反映环境污染造成的长期影响[6], 同时发现浮游细菌群落的组成差异与有机污染程度显著相关(图 2).因此, 提出利用敏感的浮游细菌作为评价有机污染水平的生物指标.采用指示值法筛选出35个指示细菌种(OTUs水平), 各指示种的相对丰度均与有机污染浓度显著相关(P < 0.05)(图 5), 且各指示种的变化与其已知的生态功能一致.这些指示种主要来源于α-变形菌纲(立克次氏体、红杆菌科)、浮霉菌门和γ-变形菌纲.立克次氏体的相对丰度在杭州湾海域较高, 与有机污染正相关, 并可能与水产养殖病害发生有密切关系.有研究表明立克次氏体和红细菌可能导致对虾的大量死亡[35].某些分类群(包括红杆菌科、弧菌属和黄杆菌科)过多也会导致细菌群落状态从健康转为患病[36].浮霉菌门和δ-变形杆菌富集为群落中的黏附颗粒, 可能解释了这些分类群与悬浮颗粒之间的正相关性.拟杆菌为严格的厌氧菌, 自由生活并附着于有机聚集体, 与海洋植物或动物残体降解相关, 是沿海远洋栖息地中最丰富的细菌群[37].邻近外海的拟杆菌如黄杆菌科、蟑螂杆状体科较丰富.有报道表明黄杆菌具有降解复杂有机物质和纤维素、几丁质等生物聚合物的特殊能力[38, 39], 且黄杆菌科能抑制弧菌的生长繁殖, 缓减了水产养殖病害的发生, 这可能使其海水有机污染程度比杭州湾小.黄杆菌还表现出作为藻类衍生代谢物消费者的潜在作用[37].有研究表明, OM60和黄杆菌富集于硝酸盐修饰的寡营养环境中, 且OM60的相对丰度与叶绿素a含量正相关[40, 41], 这与笔者观察的结果相一致.除此之外, 酸杆菌和芽单胞杆菌是土壤中最丰富的细菌门; 因此, 从钱塘江到海岸大量陆地投入可以使这些菌在杭州湾相对丰度较高.硝化螺旋菌在杭州湾的主导地位对于生物地球化学氮循环至关重要, 而硝化细菌会随着环境(如温度、pH、溶氧量等)的不断变化而产生很大差异.此外, 未鉴定的β-变形菌相对丰度与有机污染严重程度正相关, 但缺乏物种相关信息, 目前不能推测其对生态功能的影响[42].这些敏感的细菌类群均与有机污染显著相关, 是有机污染环境的敏感响应者, 可能作为评估海水有机污染水平的生物学指标.
4 结论(1) 杭州湾海域人为活动及陆源输入导致有机污染程度增加, 导致好营养型的α-变形菌纲、γ-变形菌纲的相对丰度增加, 而寡营养型的蓝细菌和拟杆菌的相对丰度减少.
(2) 有机污染主要通过改变海水理化性质的间接作用影响浮游细菌群落组成, 其中营养盐、悬浮颗粒、盐度等差异显著, 导致浮游细菌群落组成发生变化, 此外潜在病原菌丰度增加, 可能威胁邻近海域的水产养殖.
(3) 浮游细菌群落组成差异与有机污染水平显著相关, 表明微生态系统对环境污染敏感, 可作为评价海洋有机污染水平敏感的生物学指标.
| [1] | Fodelianakis S, Papageorgiou N, Pitta P, et al. The pattern of change in the abundances of specific bacterioplankton groups is consistent across different nutrient-enriched habitats in Crete[J]. Applied and Environmental Microbiology, 2014, 80(13): 3784-3792. DOI:10.1128/AEM.00088-14 |
| [2] | Xiong J B, Chen H P, Hu C J, et al. Evidence of bacterioplankton community adaptation in response to long-term mariculture disturbance[J]. Scientific Reports, 2015, 5: 15274. DOI:10.1038/srep15274 |
| [3] | Dai T J, Zhang Y, Tang Y S, et al. Identifying the key taxonomic categories that characterize microbial community diversity using full-scale classification:a case study of microbial communities in the sediments of Hangzhou Bay[J]. FEMS Microbiology Ecology, 2016, 92(10): fiw150. DOI:10.1093/femsec/fiw150 |
| [4] | Hanson C A, Fuhrman J A, Horner-Devine M C, et al. Beyond biogeographic patterns:processes shaping the microbial landscape[J]. Nature Reviews Microbiology, 2012, 10(7): 497-506. DOI:10.1038/nrmicro2795 |
| [5] | Labbate M, Seymour J R, Lauro F, et al. Editorial:anthropogenic impacts on the microbial ecology and function of aquatic environments[J]. Frontiers in Microbiology, 2016, 7: 1044. |
| [6] | Dai W F, Zhang J J, Tu Q C, et al. Bacterioplankton assembly and interspecies interaction indicating increasing coastal eutrophication[J]. Chemosphere, 2017, 177: 317-325. DOI:10.1016/j.chemosphere.2017.03.034 |
| [7] | Smith M B, Rocha A M, Smillie C S, et al. Natural bacterial communities serve as quantitative geochemical biosensors[J]. mBio, 2015, 6(3): e00326-15. |
| [8] | Van der Gucht K, Cottenie K, Muylaert K, et al. The power of species sorting:local factors drive bacterial community composition over a wide range of spatial scales[J]. Proceedings of the National Academy of Sciences of the United States of America, 2007, 104(51): 20404-20409. DOI:10.1073/pnas.0707200104 |
| [9] |
王春忠, 林国荣, 严涛, 等. 长毛对虾海水养殖环境以及虾肠道微生物群落结构研究[J]. 水产学报, 2014, 38(5): 706-712. Wang C Z, Lin G R, Yan T, et al. Microbial community in the shrimp (Penaeus penicillatus) intestine and its culture environment[J]. Journal of Fisheries of China, 2014, 38(5): 706-712. |
| [10] |
胡常巨, 熊金波, 陈和平, 等. 象山港网箱养殖区与非养殖区的细菌群落分布[J]. 生态学报, 2015, 35(24): 8053-8061. Hu C J, Xiong J B, Chen H P, et al. Distribution of bacterioplankton communities in cage culture and non-cultured areas of Xiangshan Bay, Ningbo, China[J]. Acta Ecologica Sinica, 2015, 35(24): 8053-8061. |
| [11] |
李云, 李道季, 唐静亮, 等. 长江口及毗邻海域浮游植物的分布与变化[J]. 环境科学, 2007, 28(4): 719-729. Li Y, Li D J, Tang J L, et al. Phytoplankton distribution and variation in the Yangtze River Estuary and its adjacent sea[J]. Environmental Science, 2007, 28(4): 719-729. |
| [12] |
柴超, 俞志明, 宋秀贤, 等. 长江口水域富营养化特性的探索性数据分析[J]. 环境科学, 2007, 28(1): 53-58. Chai C, Yu Z M, Song X X, et al. Exploratory data analysis to the study of eutrophication in the Yangtze River Estuary, China[J]. Environmental Science, 2007, 28(1): 53-58. |
| [13] |
柴小平, 胡宝兰, 魏娜, 等. 杭州湾及邻近海域表层沉积物重金属的分布、来源及评价[J]. 环境科学学报, 2015, 35(12): 3906-3916. Chai X P, Hu B L, Wei N, et al. Distribution, sources and assessment of heavy metals in surface sediments of the Hangzhou Bay and its adjacent areas[J]. Acta Scientiae Circumstantiae, 2015, 35(12): 3906-3916. |
| [14] |
王新, 李志江, 郑天凌. 海洋浮游细菌在东海赤潮高发区的分布与活性[J]. 环境科学, 2010, 31(2): 287-295. Wang X, Li Z J, Zheng T L. Distribution and activity of marine bacterioplankton at frequent HAB area of East China Sea[J]. Environmental Science, 2010, 31(2): 287-295. |
| [15] | Mago Dč T, Salzberg S L. FLASH:fast length adjustment of short reads to improve genome assemblies[J]. Bioinformatics, 2011, 27(21): 2957-2963. DOI:10.1093/bioinformatics/btr507 |
| [16] | Caporaso J G, Kuczynski J, Stombaugh J, et al. QⅡME allows analysis of high-throughput community sequencing data[J]. Nature Methods, 2010, 7(5): 335-336. DOI:10.1038/nmeth.f.303 |
| [17] | Edgar R C. Search and clustering orders of magnitude faster than BLAST[J]. Bioinformatics, 2010, 26(19): 2460-2461. DOI:10.1093/bioinformatics/btq461 |
| [18] | Caporaso J G, Bittinger K, Bushman F D, et al. PyNAST:a flexible tool for aligning sequences to a template alignment[J]. Bioinformatics, 2010, 26(2): 266-267. |
| [19] | DeSantis T Z, Hugenholtz P, Larsen N, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB[J]. Applied and Environmental Microbiology, 2006, 72(7): 5069-5072. DOI:10.1128/AEM.03006-05 |
| [20] |
徐国锋, 龙绍桥, 秦铭俐, 等. 乐清湾养殖区富营养化现状分析与评价[J]. 海洋环境科学, 2009, 28(S1): 59-61. Xu G F, Long S Q, Qin M L, et al. Analysis and assessment on eutrophication status in culture zone of Yueqing Bay[J]. Marine Environmental Science, 2009, 28(S1): 59-61. |
| [21] |
刘乐冕, 杨军, 余小青, 等. 厦门后溪流域沿城乡梯度浮游细菌多样性及其与环境因子的关系[J]. 应用与环境生物学报, 2012, 18(4): 591-598. Liu L M, Yang J, Yu X Q, et al. Bacterioplankton diversity in Xiamen Houxi River watershed along an urban-rural gradient and its relation to environmental factors[J]. Chinese Journal of Applied and Environmental Biology, 2012, 18(4): 591-598. |
| [22] | De'ath G. Multivariate regression trees:a new technique for modeling species-environment relationships[J]. Ecology, 2002, 83(4): 1105-1117. |
| [23] | Ramette A, Tiedje J M. Multiscale responses of microbial life to spatial distance and environmental heterogeneity in a patchy ecosystem[J]. Proceedings of the National Academy of Sciences of the United States of America, 2007, 104(8): 2761-2766. DOI:10.1073/pnas.0610671104 |
| [24] | Murray A G, Jackson G A. Viral dynamics:a model of the effects of size, shape, motion and abundance of single-celled planktonic organisms and other particles[J]. Marine Ecology Progress Series, 1992, 89: 103-116. DOI:10.3354/meps089103 |
| [25] | Cottrell M T, Kirchman D L. Community composition of marine bacterioplankton determined by 16S rRNA gene clone libraries and fluorescence in situ hybridization[J]. Applied and Environmental Microbiology, 2000, 66(12): 5116-5122. DOI:10.1128/AEM.66.12.5116-5122.2000 |
| [26] | Fernández-Gómez B, Richter M, Schüler M, et al. Ecology of marine Bacteroidetes:a comparative genomics approach[J]. ISME Journal, 2013, 7(5): 1026-1037. DOI:10.1038/ismej.2012.169 |
| [27] | Ganesh S, Parris D J, DeLong E F, et al. Metagenomic analysis of size-fractionated picoplankton in a marine oxygen minimum zone[J]. ISME Journal, 2014, 8(1): 187-211. DOI:10.1038/ismej.2013.144 |
| [28] |
高生泉, 陈建芳, 金海燕, 等. 杭州湾及邻近水域营养盐的时空分布与富营养化特征[J]. 海洋学研究, 2011, 29(3): 36-47. Gao S Q, Chen J F, Jin H Y, et al. Characteristics of nutrients and eutrophication in the Hangzhou Bay and its adjacent waters[J]. Journal of Marine Sciences, 2011, 29(3): 36-47. |
| [29] | Jiao S, Chen W M, Wang E T, et al. Microbial succession in response to pollutants in batch-enrichment culture[J]. Scientific Reports, 2016, 6: 21791. DOI:10.1038/srep21791 |
| [30] |
杨季芳, 王海丽, 陈福生, 等. 象山港海域细菌的分布特征及其环境影响因素[J]. 生态学报, 2011, 31(14): 4007-4018. Yang J F, Wang H L, Chen F S, et al. Distribution of marine bacteria and their environmental factors in Xiangshan Bay[J]. Acta Ecologica Sinica, 2011, 31(14): 4007-4018. |
| [31] | Lozupone C A, Knight R. Global patterns in bacterial diversity[J]. Proceedings of the National Academy of Sciences of the United States of America, 2007, 104(27): 11436-11440. DOI:10.1073/pnas.0611525104 |
| [32] | Bouvier T C, del Giorgio P A. Compositional changes in free-living bacterial communities along a salinity gradient in two temperate estuaries[J]. Limnology and Oceanography, 2002, 47(2): 453-470. DOI:10.4319/lo.2002.47.2.0453 |
| [33] | Cottrell M T, Kirchman D L. Contribution of major bacterial groups to bacterial biomass production (thymidine and leucine incorporation) in the Delaware estuary[J]. Limnology and Oceanography, 2003, 48(1): 168-178. DOI:10.4319/lo.2003.48.1.0168 |
| [34] | Fortunato C S. Spatial and temporal variability of bacterioplankton communities across river to ocean environmental gradients[D]. College Park, MD: University of Maryland, 2012. |
| [35] | Zhang D M, Wang X, Xiong J B, et al. Bacterioplankton assemblages as biological indicators of shrimp health status[J]. Ecological Indicators, 2014, 38: 218-224. DOI:10.1016/j.ecolind.2013.11.002 |
| [36] | Xiong J B, Zhu J Y, Dai W F, et al. Integrating gut microbiota immaturity and disease-discriminatory taxa to diagnose the initiation and severity of shrimp disease[J]. Environmental Microbiology, 2017, 19(4): 1490-1501. DOI:10.1111/1462-2920.13701 |
| [37] | Alonso C, Warnecke F, Amann R, et al. High local and global diversity of Flavobacteria in marine plankton[J]. Environmental Microbiology, 2007, 9(5): 1253-1266. DOI:10.1111/emi.2007.9.issue-5 |
| [38] | Kirchman D L. The ecology of Cytophaga-Flavobacteria in aquatic environments[J]. FEMS Microbiology Ecology, 2002, 39(2): 91-100. |
| [39] | Williams T J, Wilkins D, Long E, et al. The role of planktonic Flavobacteria in processing algal organic matter in coastal East Antarctica revealed using metagenomics and metaproteomics[J]. Environmental Microbiology, 2013, 15(5): 1302-1317. DOI:10.1111/emi.2013.15.issue-5 |
| [40] | Goldberg S J, Nelson C E, Viviani D A, et al. Cascading influence of inorganic nitrogen sources on DOM production, composition, lability and microbial community structure in the open ocean[J]. Environmental Microbiology, 2017, 19(9): 3450-3464. DOI:10.1111/emi.2017.19.issue-9 |
| [41] | Yan S, Fuchs B M, Lenk S, et al. Biogeography and phylogeny of the NOR5/OM60 clade of Gammaproteobacteria[J]. Systematic and Applied Microbiology, 2009, 32(2): 124-139. DOI:10.1016/j.syapm.2008.12.001 |
| [42] |
戴文芳, 郭永豪, 郁维娜, 等. 三门湾近海有机污染对浮游细菌群落的影响[J]. 环境科学, 2017, 38(4): 1414-1422. Dai W F, Guo Y H, Yu W N, et al. Effects of coastal organic pollution on bacterioplankton community in Sanmen Bay[J]. Environmental Science, 2017, 38(4): 1414-1422. |