 2024, Vol. 45
2024, Vol. 45

2. 中国气象局-南开大学大气环境与健康研究联合实验室,天津 300074;
3. 南开大学环境科学与工程学院,天津市城市交通污染防治研究重点实验室,天津 300071;
4. 中国科学院大气物理研究所大气边界层物理和大气化学国家重点实验室,北京 100029;
5. 中国科学院大气物理研究所国际气候与环境科学中心,北京 100029;
6. 中国科学院大学地球与行星科学学院,北京 100049
 , HAN Su-qin1,2
, HAN Su-qin1,2 , LIU Ke-xin3 , TANG Xiao4 , KONG Lei4 , DING Jing1,2 , FAN Wen-yan1,2 , WANG Zi-fa4,5,6
, LIU Ke-xin3 , TANG Xiao4 , KONG Lei4 , DING Jing1,2 , FAN Wen-yan1,2 , WANG Zi-fa4,5,6
2. Cooperative Laboratory for Atmospheric Environment-Health Research, China Meteorological Administration-Nankai University(CMA-NKU), Tianjin 300074, China;
3. Tianjin Key Laboratory of Urban Transport Emission Research, College of Environmental Science and Engineering, Nankai University, Tianjin 300071, China;
4. State Key Laboratory of Atmospheric Boundary Layer Physics and Atmospheric Chemistry(LAPC), Institute of Atmospheric Physics, Chinese Academy of Sciences, Beijing 100029, China;
5. International Center for Climate and Environment Science, Institute of Atmospheric Physics, Chinese Academy of Sciences, Beijing 100029, China;
6. College of Earth and Planetary Sciences, University of Chinese Academy of Sciences, Beijing 100049, China
中国北方城市秋冬季仍频繁经历霾天气, 无机气溶胶(SIA)是导致大气污染形成的关键气溶胶[1]. 此外, SIA会增加土壤酸度[2], 降低大气能见度[3], 影响大气辐射平衡[4]. 长期暴露在高浓度SIA的环境中有损于人类寿命和公众健康[5]. 因此, 探究污染期间SIA成因及来源具有重要意义.
无机气溶胶包含硫酸盐、硝酸盐和铵盐, 主要是直接排放到大气中的气态前体物SO2和NOx经过一系列氧化反应生成酸性物质, 再与NH3等碱性物质发生中和反应生成的. 前体物浓度是SIA组分生成的首要因素[6]. 2014~2017年, 由于煤炭燃烧的控制和能源结构的优化, 北京SO2排放快速减少, 导致硫酸盐浓度降低[7]. 北京地区这种“贫硫”条件是硝酸盐浓度及其在PM2.5中占比增加的重要原因[8]. 1990~2005年期间NH3排放量增加了90%, 导致硝酸盐和硫酸盐气溶胶浓度增加了约50%~60%[9]. SIA二次生成的化学反应包括气相、液相和非均相反应, 不同反应的重要性有所不同. Liu等[10]根据河南省2017~2018年的观测数据研究表明, 均相和非均相过程对硝酸盐的生成均起到重要作用, 而非均相过程是硫酸盐生成的主要过程. 在冬季是SIA快速增长期, 非均相过程可能对华北地区二次无机气溶胶生成起决定性作用[11, 12]. 然而, Lu等[13]基于北京地区综合观测试验发现, 冬季霾污染期间大气氧化剂浓度高于夏季, 活跃和高效的光化学对二次污染物的形成至关重要.
气象因素也是影响二次无机组分生成的关键因子. 温度可以影响无机气溶胶的非均相生成, N2O5被气溶胶表面摄取的能力与温度存在着明显的反相关关系[14]. 在高密度水汽存在时, NO2在海盐气溶胶表面上的非均相反应速率加快[15]. Zhao等[16]的研究发现, 雾-霾日相对湿度升高促进SO2的液相氧化反应的发生. 北京地区冬季污染期间, 硫酸盐浓度在低相对湿度呈下降趋势, 在高相对湿度下显著增加, 增长率为0.81 µg·(m3·h)-1, 而硝酸盐在高、低相对湿度下生长速率相似[17]. 目前定性分析气象要素对无机气溶胶影响的研究较多, 而评估高浓度无机气溶胶与重要气象因子的量化关系较少[18].
此外, 区域输送也会造成城市大气污染的发生以及无机气溶胶浓度的快速增长. 在冬季重霾事件中, 外来源地成为北京和上海污染过程的关键因素, 贡献率分别为60%[11]和45%[19]. Yang等[20]的研究结果表明, 局部化学转化不能完全解释短时间内二次无机气溶胶浓度的快速增加, 而区域传输对高浓度二次无机气溶胶的贡献至关重要. 后向轨迹模式是研究污染物区域输送常用工具之一, 原理简单, 且易操作, 结果较为直观[21]. 大气化学传输模式能够模拟大气污染物的三维时空演变特征, 从物理和化学机制上解析污染成因和来源, 是科学研究和管理决策的重要工具. 基于大气化学传输模式的源追踪法也是区域输送研究的重要方法. 与后向轨迹模式相比, 它可以同时定量评估不同标记源区对多种大气污染物的浓度贡献. 目前, 国内外常见的如CAMx模式中的在线颗粒物源识别技术PSAT[22], CMAQ耦合的ISAM模块[23]、NAQPMS中在线污染物来源追踪方法[24]等. Wang等[25]利用WRF-CAMx研究北京两类污染事件时发现, 局地排放对两个污染过程累积阶段PM2.5分别起到降低和增加的作用, 对两类事件PM2.5浓度的平均贡献率为47.3%和77.1%. Lu等[26]利用耦合了在线污染物来源追踪方法的NAQPMS探究了2014年1月武汉PM2.5时间演变成因发现, 华北平原地区污染气团的远距离输送是导致武汉PM2.5浓度急剧上升的驱动因素. Wang等[27]基于CMAQ模式量化了河北省二次无机气溶胶来源, 结果发现, 石家庄、邢台和邯郸二次无机组分总浓度的外来源贡献率分别为40.9%、62.0%和59.1%. Wang等[28]解析了上海市秋季一次污染过程细颗粒物及其组分来源, 结果表明, 与元素碳相比, 区域输送对3类无机盐的贡献更大. 外来源输送对长三角地区硝酸盐贡献率甚至达到60%~98%[29].
天津是京津冀城市圈的工业城市, 其拥有的石化、能源、装备、轮船航运和石油开采等重工业产业向大气中排放大量的大气污染物, 导致过去十几年间频繁发生霾污染事件, 特别是冬季[30], 污染期间二次无机离子是PM2.5的关键组分[31, 32]. 前期涉及天津地区细颗粒物/无机气溶胶的研究大多围绕污染特征、行业来源[30, 32~35]及区域来源[36, 37]等. 然而, 天津污染过程期间无机气溶胶区域来源及形成机制仍需要大量的综合性研究. 2020年1月, 天津共经历15个污染天, 本研究挑选了其中典型高浓度无机气溶胶过程, 结合观测数据分析和数值模拟, 综合分析气象因素、区域传输和化学过程对无机气溶胶生成的影响, 以期为理解霾天气关键组分成因提供科学依据.
1 材料与方法 1.1 观测数据2020年1月天津市主要的气象要素逐时数据, 包括地面2 m温度、2 m相对湿度、10 m风向和10 m风速来自天津市气象局, 用于验证数值模式模拟的气象场时空分布特征, 分析气象条件对无机气溶胶浓度演变过程的影响. 为探究PM2.5各组分特征, 评估大气化学传输模式模拟能力, 也获取了2020年1月天津PM2.5及其组分, 包括有机碳(OC)、元素碳(EC)、硫酸盐(SO42-)、硝酸盐(NO3-)、铵盐(NH4+)和HONO逐时数据.
采样点位于天津市津南区南开大学大气环境综合观测站(38°59′40″N, 117°20′06″E), 采样时间为2020年1月1~31日. 采用在线离子色谱仪(AIM-URG9000D, URG Corporation)对PM2.5中水溶性离子的质量浓度进行监测. 该仪器主要采用水蒸气喷射采样技术的气体/气溶胶自动采样装置(AIM)和离子色谱系统(ICS-1100)组成. 该系统具有较低的检出限(0.001 μg·m-3). 本实验所用标准溶液均为优级纯, 每月重新配制标准溶液, 绘制的标准曲线相关系数除NH4+外, 均大于99.9%(NH4+相关系数大于99.5%). 定期检查AIM系统采样口处的流量(3 L·min-1). 采用聚光科技有限公司生产的在线OC/EC分析仪(OCEC-100)对有机碳和元素碳进行在线监测, 该仪器是根据OC和EC在不同温度下的氧化顺序对其进行分离, 并基于热光透射法和热光反射法的基本原理进行测量. 数据质量控制主要包括检查采样数据连续性和数值大小, 基于天津地区前期研究, 判断大气污染物浓度数值大小是否处于正常范围, 对存疑数据结合PM2.5时间序列进行核实等方法对数据进行质量控制.
1.2 大气化学传输模式及输入数据研究采用中国科学院大气物理研究所开发的嵌套网格空气质量模式(nested air quality prediction modeling system, NAQPMS)[38], 其空间结构为三维欧拉输送模型, 采用地形追随坐标作为垂直坐标, 可以同时模拟PM10、PM2.5、SO2、NOx、CO、O3和NH3等多种污染物. NAQPMS包含污染物排放、平流输送、湍流扩散、干湿沉降、气相、液相及非均相反应等多种物理和化学过程. 其中, 干沉降的模拟采用Wesely阻力模型[39, 40], 湿沉降和液相化学采用了基于RADM的模式改进方案[41], 气相化学采用CBM-Z碳键反应机制[42], 该机制具有134个核心化学反应. 对于气溶胶过程, NAQPMS使用气溶胶热力学模块ISORROPIA1.7来处理硫酸盐、硝酸盐和铵盐的气粒分配和热力学平衡[43]. 为考虑气体和气溶胶之间的相互作用, NAQPMS考虑了14种化合物和28种非均相反应, 气溶胶介质包括硫酸盐、黑碳、沙尘和海盐等[44]. 输入NAQPMS模式的气象数据由WRF3.9(weather research and forecasting model version3.9)提供[45]. WRF是由美国的National Center for Environmental Prediction(NCEP)和National Center for Atmospheric Research(NCAR)等多家机构联合开发和维护的中尺度数值天气预报系统. WRF模式提供了主要包括长波RRTM[46]/短波辐射模块Gorddard方案[47]、陆面过程Noah方案[48]、积云对流Grell 3 d方案[49]、边界层湍流Mellor-Yamada-Janjic方案[50]和Lin云微物理[51]等一系列物理机制和参数化方案.
WRF设置双层嵌套模拟区域(D01和D02), D01包含东亚大部分地区, D02涵盖中国地区, 水平分辨率分别为15 km和5 km, 垂直方向共20层. WRF模拟方案采取运行36 h, 取后24 h模拟数据作为NAQPMS气象驱动场的模拟方式, 其初始条件和边界条件由NCEP/NCAR提供的1° × 1°再分析资料(FNL)提供. NAQPMS的化学边界条件由全球大气化学传输模式MOZART提供[52, 53], 前15 d模拟作为NAQPMS的spinup时间. 此外, 输入NAQPMS的排放清单包括HTAPv2.2全球人为源清单[54]、GFED4生物质燃烧排放清单[55, 56]、MEGAN-MACC生物VOC源排放清单[57]等, 涉及SO2、NOx、CO、NMVOC(非甲烷挥发性有机化合物)、NH3、PM10、PM2.5、BC、OC和CO2等多物种的排放.
为量化天津地区SIA的区域来源, 本研究采用耦合在NAQPMS中的在线污染物来源追踪方法量化标记出的污染源区对目标城市多种大气污染物的贡献[24, 58], 分析区域传输对所研究地区污染物浓度的影响. 这一方法能够同时考虑排放、物理(包含平流、扩散和对流等)和化学等多种大气过程, 计算得到不同区域对目标城市一次和二次气溶胶浓度的贡献. 污染物根据省市行政区进行划分, 每一个定义的源地的贡献在模块中是正值. 基于地理位置, 计算了5个定义区域(见图 1), 包括天津、河北+北京、山东、河南及其它地区排放对天津SIA浓度的贡献.

|
图 1 在线污染物追踪方法标记的源区 Fig. 1 Tagged source regions used by an online source-tagging module |
为综合评估WRF-NAQPMS的模拟能力, 首先对比观测的和模拟的2 m温度(T2)、2 m相对湿度(RH2)、10 m风速(WS10)小时数据(如图 2), 计算相关系数(R)、均方根误差(RMSE)、平均偏差(MB)和平均误差(ME), 见公式(1)~(4):
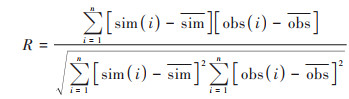
|
(1) |
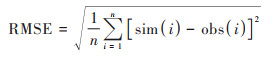
|
(2) |

|
(3) |
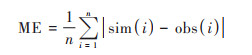
|
(4) |

|
图 2 观测的和模拟的2 m温度(T2)、2 m相对湿度(RH2)和10 m风速(WS10)时间序列对比 Fig. 2 Comparison of observed and simulated 2 m temperature (T2), 2 m relative humidity (RH2), and 10 m wind speed (WS10) |
式中, sim为模拟值, obs为观测值, n表示有效数据对. 结果发现, WRF对温度模拟效果最好, 整个研究时段温度小时观测值和模拟值的日变化较为一致, 相关系数达到0.92, 平均偏差为-0.8℃, 均方根误差为2.2℃;其次是相对湿度, 观测和模拟值的相关系数为0.73, 模式整体表现稍有低估, 主要是在一些个别高湿时段. WRF对风速略有高估, 平均偏差为1.2 m·s-1, 均方根误差为1.7 m·s-1, 但整体上能再现风速的时间演变, 相关系数达到0.5, 特别在较强风速时段. 因此, WRF在本研究模式设置下对天津地区主要气象要素具有较好的模拟性能, 可为NAQPMS提供稳定可靠的气象数据.
其次, 如图 3所示的无机气溶胶对比结果发现, 观测和模拟的硫酸盐、硝酸盐和铵盐随时间呈现出较为一致的趋势, 模式对铵盐的模拟效果最好, 相关系数为0.73, 平均偏差和平均误差均最小, 分别为-3.7 μg·m-3和7 μg·m-3, 略有低估. 同样, NAQPMS也能较好地再现硝酸盐和硫酸盐的时间演变特征, 相关系数分别为0.67和0.61, 平均偏差分别为5.3 μg·m-3和1 μg·m-3, 模式略有高估. 整体上, NAQPMS对两次典型污染过程的无机组分都有较好的模拟性能, 为本研究追踪无机气溶胶的来源提供可靠的数据基础.

|
图 3 观测的和模拟的硝酸盐(NO3-)、硫酸盐(SO42-)和铵盐(NH4+)小时浓度对比 Fig. 3 Comparison of hourly observed and simulated nitrate (NO3-), sulfate (SO42-), and ammonium (NH4+) concentrations |
基于地面观测数据, 天津2020年1月月均ρ(PM2.5)值为99.2 μg·m-3, 远超过PM2.5环境空气质量二级标准(日均值> 75 μg·m-3), 也高于2018年(56.55 μg·m-3)和2019年(97.51 μg·m-3)同期PM2.5浓度水平[32]. ρ(SIA)月均值为36.1 μg·m-3, 是ρ(OC)月均值(7.4 μg·m-3)的4.9倍, ρ(SIA)最高日均值和小时值分别为105.1 μg·m-3和132.1 μg·m-3. SIA中硝酸盐浓度最大, ρ(NO3-)月均值为18.2 μg·m-3, 是ρ(SO42-)(9.2 μg·m-3)和ρ(NH4+)(8.7 μg·m-3)月均值的2倍. 根据表 1中显示的已有的研究结果, 2017年1月, 天津硫酸盐浓度水平略高于硝酸盐, 自2018年开始, 硝酸盐赶超硫酸盐, 导致天津在冬季月份由硫酸盐污染转变为硝酸盐污染, 这主要是2017年开始中国北方“2+26”城市大力实施“代煤工程”所导致的[59].
|
|
表 1 2017~2020年1月天津PM2.5无机组分浓度/μg·m-3 Table 1 Concentrations of inorganic aerosol species in PM2.5 in January from 2017 to 2020/μg·m-3 |
将PM2.5日均浓度超过75 μg·m-3定义为污染天, 2020年1月天津共出现15个污染天, 为保证更多的样本量, 将连续3 d及以上污染天设定为一个污染过程, 2020年1月共有3个过程, 选择SIA浓度均值最高的两次1月15~19日, 1月26~28日作为本研究分析过程, CASE1和CASE2时段划分见图 4. 根据表 2观测数据统计情况, CASE1污染天持续5 d, ρ(SIA)均值更高, 为76.8 μg·m-3;CASE2持续3 d, ρ(SIA)均值为66.0 μg·m-3, CASE1中SIA各组分绝对浓度均高于CASE2. ρ(SIA)日均和小时峰值分别为105.1 μg·m-3和132.1 μg·m-3, 出现在CASE1的1月17日. 两个过程中ρ(NO3-)分别为37.0 μg·m-3和31.4 μg·m-3, 浓度最高, 其次为ρ(SO42-), 分别为21.0 μg·m-3和19.6 μg·m-3, NH4+浓度最低. 此外, CASE1过程ρ(NO2)为71.5 μg·m-3, 是CASE2(49.6 μg·m-3)的1.4倍, ρ(SO2)和ρ(NH3)均值分别为7.9 μg·m-3和13.5 μg·m-3, 略低于CASE2过程的两种气体浓度(8.7 μg·m-3和14.4 μg·m-3). 综上, 与CASE2相比, CASE1污染过程持续时间长, 无机气溶胶总量及其各组分浓度更高, 但主要前体物中只有NO2浓度更高, SO2和NH3浓度略低. 这表明前体物不是高浓度无机气溶胶的决定性因素, 二次无机组分的生成存在强的非线性特征.

|
图 4 天津风场、相对湿度、无机气溶胶总量(SIA)及各组分、SO2、NO2、有机碳(OC)和元素碳(EC)小时值时间演变 Fig. 4 Hourly variation in wind, relative humidity, SIA, SIA components, SO2, NO2, OC, and EC over Tianjin |
|
|
表 2 天津两个污染过程SIA及各组分、前体物浓度和气象要素特征 Table 2 General information of SIA, SIA components, precursor concentration, and meteorological elements in the two polluted processes over Tianjin |
图 4显示了两个过程风场、相对湿度和PM2.5组分及气体随时间的演变(污染物不连续处为观测数据缺省), CASE1过程前期(1月15日), 在近地面弱风控制下, SIA浓度呈现波动式快速增加的趋势;1月16~17日为污染过程稳定期, SIA总量保持高浓度值;1月18~19日, 较强西南风和高湿条件(> 80%)持续控制下, SIA浓度逐渐下降, 持续性较强西南风多数情况下受大尺度系统控制;1月19日夜间至20日凌晨, 在干燥的强西南风转强北风的驱动下, 污染过程结束. 整个过程硫酸盐、硝酸盐和铵盐随时间的变化基本一致, 硝酸盐浓度均高于其它两种无机气溶胶, 特别是在污染维持阶段, 差值可达5~25 μg·m-3. NO2和SO2在SIA快速增加之前(1月14日)已出现并持续保持高浓度水平, 为污染过程无机气溶胶的快速生成提供前体物.
CASE2过程高浓度SIA时段, 近地面大气基本处于相对湿度 > 60%条件下, 风速小, 风向为偏北风和南风交替出现, 多变的风向易产生局地辐合风场, 不利于大气污染物的扩散. CASE2前期(1月25~26日), ρ(SIA)逐渐升高形成第一个峰值, 为104.7 μg·m-3, 随后SIA浓度短时下降后再缓慢增加, 于1月28日06:00形成次峰值, 浓度为102.7 μg·m-3, CASE2后期(1月28~30日)SIA浓度缓慢下降, 污染过程结束. 与CASE1相似, 3种无机气溶胶变化趋势基本一致, 硝酸盐整体高于硫酸盐和铵盐. 但CASE2过程SIA第一个峰值时刻, 硫酸盐浓度最高. 较清洁时段(例如1月的20日和24日), CASE2过程中NO2在微小波动中整体保持高浓度水平;SO2呈现日变化特征, 每日日间出现峰值, 主要受工业排放规律的影响, 这与CASE1存在区域性阶段不同, 侧面体现了CASE2的局地性特征. 两个过程EC浓度差异不大, 但CASE2过程OC浓度较高, 且变化趋势与SIA更为一致, 也成为PM2.5污染的重要组分.
进一步了解天津冬季两个无机气溶胶总量及其各组分、主要气象要素的日变化特征, 如图 5所示. SIA总量与其各组分日变化规律基本保持一致, 但CASE1和CASE2之间存在差异. CASE1过程11:00 SIA与3类气溶胶同步出现一个明显峰值, 这可能主要与日间排放的大量前体物气相氧化有关. 午后无机气溶胶浓度下降, 16:00出现最低值, 这是由于午后温度升高, 相对湿度降低, 风速增加至16:00达到峰值, 转好的边界层内扩散条件促使污染物浓度下降. 16:00至夜间SIA浓度整体呈现逐渐上升趋势, 夜间高浓度无机气溶胶除受较低的边界层高度影响外, 气体的非均相转化至关重要, 这与北京地区冬季霾过程的结论较为一致[60]. CASE2过程, SIA出现弱的双峰值现象, 分别出现在10:00和17:00, 直接导致日间SIA浓度高于夜间. 前一峰值出现的原因与CASE1相似, 受边界层高度和气体的气相过程的影响大, 午后转好的扩散条件未能有效降低SIA浓度, 后一峰值的形成主要与前体物的气相转化有关. CASE2温度、相对湿度和风速趋势与CASE1基本一致, 但波动更小, 风速小于CASE1, 大气条件更趋于静稳, 局地性更强. 硫酸盐和铵盐与SIA日变化一致, 硝酸盐浓度自凌晨至17:00均保持稳定趋势, 于18:00与其它组分同步下降.

|
图 5 CASE1和CASE2过程SIA及其各组分和气象要素日变化特征 Fig. 5 Diurnal variation in SIA, SIA chemical components, and meteorological parameters in CASE1 and CASE2 |
根据上述分析, SO2和NO2是无机气溶胶污染形成的必要条件, 但不是决定性因素. 而NH3是影响硫酸盐和硝酸盐存在形式及浓度的重要碱性气体, NH3通常会优先与大气中的气态H2SO4结合生成(NH4)2SO4或NH4HSO4, 或是NH3直接在酸性颗粒物表面反应形成NH4+[61], 富余的NH3再与HNO3和HCl反应, 形成NH4NO3和NH4Cl. 为了解两个过程NH4+对SO42-和NO3-的中和程度, 图 6比较了NH4+摩尔浓度与(2SO42-+NO3-)摩尔浓度在两个过程的相关性. 发现两者之间存在很好的线性相关关系, 相关系数R在两个过程均接近1, 拟合斜率都超过1, 分别为1.03和1.12, 大气中含有充足的NH3能够完全中和H2SO4和HNO3, 硫酸盐和硝酸盐均以(NH4)2SO4和NH4NO3的形式存在, 即天津在冬季处于富氨的条件下, 与贫氨条件相比, 会生成更多的硝酸盐. 硫酸铵化学性质稳定, 而硝酸铵生成NH3和HNO3是一个可逆反应, 主要受温度的影响, 高温条件下, 硝酸铵易于分解;在冬季这种低温条件下则倾向以颗粒态硝酸铵的形式存在[62]. 此外, 两个过程ρ(HNO3)均值低, 分别为1.01 μg·m-3和0.39 μg·m-3, 也印证了生成的H2SO4和HNO3几乎被完全中和的这一结论. 但是, 研究显示富氨环境有利于SO2的非均相氧化过程, 从而促进SO42-的持续生成[63], 这或可解释两个过程中SO2浓度仅仅是NO2浓度的11%和17.5%, 但硫酸盐浓度可以达到硝酸盐的56.8%和62.4%这一情形.

|
图 6 CASE1和CASE2过程NH4+与(2SO42 -+NO3-)摩尔浓度散点分布 Fig. 6 Scatter plot of NH4+ molar concentration and (2SO42-+NO3-) molar concentration during CASE1 and CASE2 |
基于公式(5)和公式(6)计算出硫氧化率(sulfur oxidation rate, SOR)和氮氧化率(Nitrogen oxidation rate, NOR), 评估两个过程SO2和NO2分别转化为硫酸盐和硝酸盐的能力:
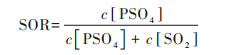
|
(5) |
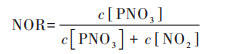
|
(6) |
式中, c为污染物的量浓度, SOR和NOR值越高表示SO2和NO2越高的氧化效率, 意味着大气中生成更多二次无机气溶胶[64]. 两个过程SOR值相差不大, 分别为0.69和0.72, 高于北京在相对湿度在70%~80%这一范围内的均值0.34[65], 和北京在霾天内SOR为0.24和0.29[66, 16], 天津在2020年冬季污染过程硫酸盐具有更高的生成能力. 两个过程的NOR均值分别为0.33和0.38, 基本为同过程SOR的一半, 硫酸盐生成能力显著高于硝酸盐.
气象因子影响硫酸盐和硝酸盐的生成能力, 图 7显示了两个过程温度/相对湿度与SOR/NOR的关系. CASE1过程SOR随相对湿度先缓慢增加后快速增加, 转折点大致在RH = 60%. 而NOR在RH < 50%时随着相对湿度的增加而降低, RH > 50%, NOR随之增加, RH在40%~60%范围内不利于硝酸盐的生成. 而温度在-10~0℃范围内对应较高的SOR和NOR, 可分别达到0.8和0.4以上. CASE2过程SOR随相对湿度的增加而增加, 但与CASE1不同的是, 增加幅度减小, 转折点在RH = 70%;而高SOR(> 0.8)对应在-5~5℃温度范围内. NOR与CASE1差异较大, 随相对湿度持续降低, 较高的NOR值(> 0.4)对应[0℃, 10℃]温度区间和[40%, 60%]相对湿度区间.

|
图 7 CASE1和CASE2过程SOR和NOR与相对湿度(RH2)及温度(T2)散点图及趋势线 Fig. 7 Scatter plot of SOR, NOR and temperature (T2), relative humidity (RH2) during CASE1 and CASE2 |
温度和相对湿度条件会影响硫酸盐和硝酸盐的生成能力, 但两个过程的SOR和NOR差异不大, 进一步量化气象参数对高浓度无机气溶胶的关系, 可以为优化大气化学传输模式中理化过程参数化方案中气象约束条件、大气污染统计预报、无机气溶胶的烟雾箱试验提供参考[18]. 由于硫酸盐和硝酸盐分别与相同温度和相对湿度区间的趋势基本一致, 此处不再一一列举. 如图 8显示的基于观测数据统计方法构建的不同温度和相对湿度区间与SIA浓度的箱式图, 结果发现, CASE1过程SIA浓度随温度变化存在两个峰值, ρ(SIA) > 80 μg·m-3(高浓度)对应[-6℃, 0℃]和[2℃, 4℃]两个温度区间. 随着相对湿度的增加, SIA浓度整体上呈现上升趋势, 但在[50%, 60%]以及[80%, 100%]两个区间对应高浓度SIA. CASE2过程, 在不同温度区间中SIA存在一个峰值, 对应着温度区间为[2℃, 4℃]. 与CASE1有所不同, CASE2过程SIA随着相对湿度的增加呈现先增加后降低的趋势, SIA浓度峰值处于[60%, 70%]相对湿度区间. 两个过程温度和相对湿度阈值的差异将在2.3节阐释. 此外, 这一结果与Han等[18]的研究结论有所差异, 2013年1月北京地区相对湿度越高, 无机气溶胶浓度越高;硫酸盐和硝酸盐随着温度的变化基本呈现正态分布的态势, 温度在-4~-6 ℃之间更适宜硫酸盐和硝酸盐的二次生成. 这表明不同城市前体物排放、气象特征和大气氧化性都可能是导致结果不同的原因.

|
灰色加号和红色直线分别为不同温度和相对湿度区间无机气溶胶(SIA)的散点图和均值趋势线 图 8 CASE1和CASE2过程无机气溶胶(SIA)在不同温度和相对湿度区间的箱式图 Fig. 8 Relationship between SIA concentrations and temperature (T2), relative humidity (RH2) in CASE1 and CASE2 |
基于污染特征分析, 两个过程风场存在区域性和局地性的差异, 可能会导致SIA的主导来源不同, 利用NAQPMS耦合的在线污染物追踪方法量化了周边地区对天津两个污染过程SIA及硫酸盐、硝酸盐浓度的贡献, 见表 3和图 9. 结果发现, CASE1过程, 天津SIA以外来源均为主导, 贡献率为62.3%, 日均贡献率最大值达到75.6%, 发生在1月18日. 外来源中北京和河北地区的贡献率最大, 为57.6%;本地排放平均贡献率为37.7%, 范围在24%~58%之间, 仅在1月17日本地贡献最大. 整个过程在SIA快速增加和快速下降阶段, 来自北京和河北地区污染物的区域输送是主要来源. 与CASE1不同, CASE2是本地排放占主导的污染过程, 平均贡献率为77.9%, 日最高贡献率为88.5%, 发生在1月28日. 外来源中也是北京和河北贡献率最大, 均值为17.9%, 最大贡献率为28.5%, 出现在1月26日. 综上, 根据天津地区的SIA的主导来源可以将CASE1和CASE2分别划分为本地源主导型和外来源主导型污染过程. 两个过程SIA在快速上升和下降阶段, 外来源贡献增加;在SIA稳定期间, 本地排放贡献增加. 此外, 京津冀地区的总贡献率在两个过程均已超过95%, 这表明城市圈内部排放是首要来源, 大气污染防控的重点可以聚焦到京津冀城市圈内.
|
|
表 3 CASE1和CASE2过程不同源区对天津SIA、硫酸盐和硝酸盐贡献率及浓度贡献1) Table 3 Contribution rates and concentration contribution of SIA, sulfate and nitrate over Tianjin from the source regions in CASE1 and CASE2 |

|
图 9 不同源区对天津地区SIA浓度贡献率及SIA日均浓度时间变化 Fig. 9 Daily average contribution rate of SIA over Tianjin from potential source regions and daily average concentration of the observed SIA |
为厘清天津地区两个过程不同阶段SIA的三维输送机制, 图 10和图 11显示了CASE1和CASE2在不同污染发展阶段SIA及水平风场的空间分布和垂直分布特征. CASE1在1月15~16日, 京津冀北部地区盛行较强北风, 污染气团向天津输送, 北风风速自北向南逐渐减小的分布态势(风速辐合)将导致传输来的和本地生成的气溶胶在天津地区发生积聚, 使得SIA浓度快速升高. 该时段本地排放和外来源输送的贡献率分别为39.8%和49.9%, 促成了污染的形成. 在垂直高度上, 同样是在偏北风控制下自北向南的输送路径, SIA在天津地区400 m高度下存在高浓度中心. 1月18日, 河北南部产生更高浓度的SIA, 在西南风的驱动下向天津输送, 导致外来源输送贡献增加, 贡献率达到73.0%;在垂直方向上, 天津西部地区500 m高度以下以西南风和偏西风为主, 污染物来源与地面保持一致. 1月19日, 京津冀地区均在较强西北风的控制下, 区域性污染天气得到缓解, 天津SIA浓度快速降低, 外来源贡献占主导, 贡献率为54.7%.

|
图 10 两个污染过程不同阶段SIA及风场空间分布随时间的演变特征 Fig. 10 Spatial distribution of SIA concentration and wind field in different stages of CASE1 and CASE2 |

|
图 11 两个污染过程不同阶段SIA及水平风场沿纬度39°6′N的垂直分布特征 Fig. 11 Vertical distribution of SIA concentration and horizontal wind at latitude 39°6′N in different stages of CASE1 and CASE2 |
CASE2过程在1月25日, 受天津以北地区较强东北风的影响, 外来源为主导来源, 贡献率为50.7%, 以京津冀北部地区的贡献为主, 该阶段与CASE1相似. 1月26日, 随着系统北风的逐渐减弱, 天津本地贡献率增加(56.7%), 成为主导来源. 在污染维持阶段, 整个京津冀地区地面基本处于静风状态, 在垂直方向上, 天津地区800 m以下高度同样为弱风条件, 本地排放贡献进一步增加, 贡献率达到88.5%. 在污染过程后期, 京津冀地区的污染气团受海上清洁气流的清除作用得以缓解, 天津地区地面和上层大气在东北风的驱动下, SIA浓度自东向西逐渐降低. 但该阶段与CASE1不同的是, 本地排放仍是SIA的主导来源, 贡献率为51.9%, 主要是因为东北风相对CASE1后期西北风风力弱, 导致外来源贡献小.
结合图 2、图 4、图 9和图 10, 阐释2.2节高浓度SIA对应的温度和相对湿度区间在两个过程的差异. CASE1过程高浓度SIA对应的温度区间比CASE2多一个低温范围, 为[-6℃, 0℃], 主要发生在CASE1污染稳定期(1月16日), 在冷的弱西北风的影响下, 北京和河北的大气污染物向天津地区输送, 贡献率为49.9%, 再加上局地生成造成无机气溶胶浓度逐渐上升. CASE1过程高浓度SIA对应的相对湿度区间比CASE2多一个的高湿时段, 为[80%, 100%], 主要发生在污染过程后期(1月18日), 在高湿的西南风的控制下, 更多的北京和河北地区排放向天津输送造成的高浓度SIA. 这一结果体现了区域输送导致两个过程高浓度SIA对应的温度和相对湿度条件的差异.
具体分析不同源地排放对天津硫酸盐和硝酸盐的贡献情况(表 3). 两个过程中硫酸盐和硝酸盐的关键来源与SIA保持一致. CASE1期间, 外来源输送是天津硫酸盐和硝酸盐的关键来源(贡献率 > 50%), 其中北京和河北的贡献率最大, 分别为47.1%和62.6%, 本地排放贡献率分别为48.1%和33.7%. CASE2过程, 本地贡献占主导, 硫酸盐和硝酸盐贡献率分别为72.8%和78.4%, 此外, 北京和河北的贡献率均 < 20%. 无机组分绝对浓度贡献结果显示, 两个过程本地排放对天津ρ(NO3-)的贡献分别为16.2 μg·m-3和29.3 μg·m-3, 均高于对天津ρ(SO42-)贡献(8.2 μg·m-3和25.1 μg·m-3), 外来输送对ρ(NO3-)贡献分别为31.7 μg·m-3和8.1 μg·m-3, 对ρ(SO42-)贡献为8.8 μg·m-3和9.4 μg·m-3, 这表明CASE1本地生成和外来源输送共同造成硝酸盐高于硫酸盐浓度, 而CASE2仅本地源造成硝酸盐浓度高于硫酸盐. 此外, 基于统计结果发现, 两个过程京津冀地区对天津硫酸盐的平均贡献率分别为95.2%和91.2%, 而对硝酸盐分别为96.4%和98.0%, 略高于硫酸盐, 这可能是因为硫酸盐寿命更长(1~2周)[67], 化学稳定性高, 而硝酸盐中重要成分硝酸铵在较强光照条件下容易分解成HNO3和NH3, 不利于长距离长时间输送.
2.4 不同化学反应对两个过程SIA生成的影响不同化学反应对两个过程SIA生成的影响可能存在差异, 借鉴Lu等[68]的研究思路, 基于NAQPMS, 利用敏感性试验方法直接和间接地量化了液相、非均相和气相过程对无机气溶胶浓度的影响, 分别为cAqueous、cHetero和cGas, 计算见公式(7)~(9):
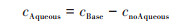
|
(7) |
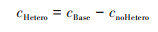
|
(8) |
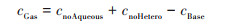
|
(9) |
式中, cBase表示NAQPMS基准模拟得到的无机气溶胶浓度, cnoAqueous和cnoHetero分别为关闭NAQPMS模式中液相过程和非均相过程模拟得到的无机气溶胶浓度. 两个过程的试验结果如图 12, 平均状态下气相过程是无机气溶胶生成的首要来源, 在CASE1和CASE2过程中贡献率分别为48.9%和57.8%;其次是非均相过程, 对SIA的贡献率分别为48.1%和42.2%;液相过程的贡献小, 但CASE1过程液相过程影响(3.0%)稍大于CASE2(0.01%).

|
图 12 液相、非均相和气相过程对天津CASE1和CASE2过程无机气溶胶总量、硫酸盐和硝酸盐的贡献率 Fig. 12 Contribution rate of aqueous, heterogeneous and gas-phase process on SIA, sulfate, and nitrate in CASE1 and CASE2 over Tianjin |
具体解析不同反应对硫酸盐和硝酸盐的影响, 两个过程硫酸盐生成的关键过程均为气相氧化过程, 贡献率相当, 分别为86.5%和86.8%. 其中, 起核心作用的气相反应主要是SO2被⋅OH等自由基的氧化反应, 见公式(10), 这一结果在前期的观测研究中也得到了证实, 冬季大气中同样存在大量的自由基和氧化剂, 促进二次无机气溶胶的光化学生成[13]. 其次是非均相过程, 贡献率分别为12.7%和13.5%, 主要来源于SO2被吸附到多种气溶胶表面, 转化为硫酸盐的反应;公式(11)为间接反应为非均相形成的HONO在日光条件下产生⋅OH自由基, 促进SO2的气相氧化反应. 非均相化学在硫酸盐生成中的重要作用已在大量的研究中得到证实[12, 69]. 两个过程中, 硝酸盐的关键过程是非均相和气相过程, CASE1非均相反应为首要来源, 贡献率为55.3%, 主要由N2O5在气溶胶表面的非均相水解反应生成, 见公式(12);气相过程的贡献率为41.2%, 主要与公式(13)这一反应有关, 光照条件下, NO2被·OH氧化产生HNO3, HNO3被碱性气体中和产生颗粒态硝酸盐. CASE2中, 气相过程为首要来源, 贡献率为53.0%, 非均相反应贡献率略低, 为47.0%. 两个过程中液相过程对硫酸盐和硝酸盐的作用均较小, 这一结果与Lu等[68]研究的结论一致, 主要是因为冬季北方城市云水含量较少, 导致无机气溶胶的液相反应影响小. 但CASE1过程的液相过程影响大于CASE2, 两个过程ρ(O3)观测值分别为15.0 μg·m-3和33.9 μg·m-3, CASE1中更低的O3浓度可能对应更弱的太阳辐射和更多的云水量, 从而有利于液相反应的发生.
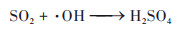
|
(10) |
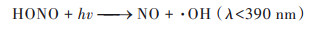
|
(11) |

|
(12) |
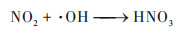
|
(13) |
两个过程不同氧化反应对硫酸盐和硝酸盐浓度贡献的时间序列如图 13(此处不再展示两个过程之外的时间段), 整体上两个过程中无机气溶胶总量贡献最大的是气相过程, 对ρ(SIA)贡献分别为30.1 μg·m-3和56.1 μg·m-3, 但在1月18日和1月27日个别时段, 非均相过程成为首要来源, 对ρ(SIA)最大贡献可达到167.7 μg·m-3. 具体而言, 硫酸盐的关键来源是气相反应, 整个时段气相过程的贡献均高于非均相和液相过程. 在两个过程的时间序列中, 硝酸盐的首要化学过程并不一致, 非均相过程为首要反应主要发生在CASE1过程中1月18日和CASE2过程1月26日凌晨、1月27日, 结合图 4中SIA与相对湿度的时间演变, 3个时段均对应持续数小时至一天的高湿环境(RH2 > 60%), 高密度水汽条件可以加快NO2在气溶胶表面上的非均相反应速率[15]. 而液相反应对整个研究时段硫酸盐和硝酸盐浓度的影响较小.

|
图 13 液相、非均相和气相过程对天津CASE1和CASE2过程无机气溶胶总量、硫酸盐和硝酸盐浓度贡献时间序列 Fig. 13 Hourly variation in concentration contribution of aqueous, heterogeneous, and gas-phase process on SIA, sulfate, and nitrate in CASE1 and CASE2 over Tianjin |
(1)天津市2020年1月共出现15个污染天, 挑选两个高浓度无机气溶胶的典型污染过程(CASE1和CASE2), 利用观测数据和耦合了在线污染物来源追踪方法的大气化学传输模式NAQPMS分析了气象因子、区域输送和化学过程对无机气溶胶生成的影响. CASE1和CASE2过程污染天分别持续5 d和3 d, ρ(SIA)均值为76.8 μg·m-3和66.0 μg·m-3, 硝酸盐浓度均高于硫酸盐和铵盐, 是典型的硝酸盐主导的污染过程. 无机气溶胶受气象因子的影响, CASE1在ρ(SIA) > 80 μg·m-3对应温度区间为[-6℃, 0℃]和[2℃, 4℃], 相对湿度区间为[50%, 60%]和[80%, 100%];CASE2过程的温度范围为[2℃, 4℃], 相对湿度范围为[60%, 70%].
(2)CASE1和CASE2过程, 外来源对天津SIA的平均贡献率分别为62.3%和22.1%, 分别为区域传输主导和局地生成主导过程. 三维SIA浓度与风场显示, 两个过程400 m高度以下不同高度的传输路径基本保持一致, 外来源主要为北京和河北地区, 但不同风向导致两个过程不同阶段来向不完全相同. 两个过程本地排放对天津ρ(NO3-)的贡献分别为16.2 μg·m-3和29.3 μg·m-3, 均高于对ρ(SO42-)贡献(8.2 μg·m-3和25.1 μg·m-3);外来输送对ρ(NO3-)贡献分别为31.7 μg·m-3和8.1 μg·m-3, 对ρ(SO42-)贡献为8.8 μg·m-3和9.4 μg·m-3, 这表明CASE1本地生成和外来源输送贡献造成硝酸盐高于硫酸盐浓度, 而CASE2仅本地源导致硝酸盐浓度高于硫酸盐.
(3)大气化学传输模式中液相、非均相和气相过程敏感性试验表明, 两个污染过程气相氧化反应是无机气溶胶生成的首要来源, 贡献率分别为48.9%和57.8%;其次是非均相过程, 贡献率分别为48.1%和42.2%. CASE1过程中硫酸盐的主要来源是气相氧化, 贡献率为86.5%, 但硝酸盐为非均相过程, 贡献率为55.3%. CASE2过程与之不同, 硫酸盐和硝酸盐的首要化学过程均为气相过程, 贡献率分别为86.8%、53.0%. 持续性高湿环境(RH2 > 60%)有利于氮氧化物在气溶胶表面上的非均相反应. 相比之下, 液相过程对无机气溶胶的贡献小.
致谢: 感谢国家重大科技基础设施项目“地球系统数值模拟装置”提供支持.
| [1] | Yang F, Tan J, Zhao Q, et al. Characteristics of PM2.5 speciation in representative megacities and across China[J]. Atmospheric Chemistry and Physics, 2011, 11(11): 5207-5219. DOI:10.5194/acp-11-5207-2011 |
| [2] | Zhao Y, Duan L, Xing J, et al. Soil acidification in China: is controlling SO2 emissions enough?[J]. Environmental Science & Technology, 2009, 43(21): 8021-8026. |
| [3] | Watson J G. Visibility: science and regulation[J]. Journal of the Air & Waste Management Association, 2002, 52(6): 628-713. |
| [4] | Crumeyrolle S, Gomes L, Tulet P, et al. Increase of the aerosol hygroscopicity by cloud processing in a mesoscale convective system: a case study from the AMMA campaign[J]. Atmospheric Chemistry and Physics, 2008, 8(23): 6907-6924. DOI:10.5194/acp-8-6907-2008 |
| [5] | Dominici F, Wang Y, Correia A W, et al. Chemical composition of fine particulate matter and life expectancy: in 95 US counties between 2002 and 2007[J]. Epidemiology, 2015, 26(4): 556-564. DOI:10.1097/EDE.0000000000000297 |
| [6] | Dong X Y, Li J, Fu J S, et al. Inorganic aerosols responses to emission changes in Yangtze River Delta, China[J]. Science of the Total Environment, 2014, 481: 522-532. DOI:10.1016/j.scitotenv.2014.02.076 |
| [7] | Li H Y, Cheng J, Zhang Q, et al. Rapid transition in winter aerosol composition in Beijing from 2014 to 2017: response to clean air actions[J]. Atmospheric Chemistry and Physics, 2019, 19(17): 11485-11499. DOI:10.5194/acp-19-11485-2019 |
| [8] | Jo Y J, Lee H J, Jo H Y, et al. Changes in inorganic aerosol compositions over the Yellow Sea area from impact of Chinese emissions mitigation[J]. Atmospheric Research, 2020, 240. DOI:10.1016/j.atmosres.2020.104948 |
| [9] | Wang S X, Xing J, Jang C, et al. Impact assessment of ammonia emissions on inorganic aerosols in East China using response surface modeling technique[J]. Environmental Science & Technology, 2011, 45(21): 9293-9300. |
| [10] | Liu H J, Tian H Z, Zhang K, et al. Seasonal variation, formation mechanisms and potential sources of PM2.5 in two typical cities in the central plains urban agglomeration, China[J]. Science of the Total Environment, 2019, 657: 657-670. DOI:10.1016/j.scitotenv.2018.12.068 |
| [11] | Li J, Du H Y, Wang Z F, et al. Rapid formation of a severe regional winter haze episode over a mega-city cluster on the North China Plain[J]. Environmental Pollution, 2017, 223: 605-615. DOI:10.1016/j.envpol.2017.01.063 |
| [12] | Zheng B, Zhang Q, Zhang Y, et al. Heterogeneous chemistry: a mechanism missing in current models to explain secondary inorganic aerosol formation during the January 2013 haze episode in North China[J]. Atmospheric Chemistry and Physics, 2015, 15(4): 2031-2049. DOI:10.5194/acp-15-2031-2015 |
| [13] | Lu K D, Fuchs H, Hofzumahaus A, et al. Fast photochemistry in wintertime haze: consequences for pollution mitigation strategies[J]. Environmental Science & Technology, 2019, 53(18): 10676-10684. |
| [14] | Escorcia E N, Sjostedt S J, Abbatt J P D. Kinetics of N2O5 hydrolysis on secondary organic aerosol and mixed ammonium bisulfate-secondary organic aerosol particles[J]. The Journal of Physical Chemistry A, 2010, 114(50): 13113-13121. DOI:10.1021/jp107721v |
| [15] | Peters S J, Ewing G E. Reaction of NO2(g) with NaCl (100)[J]. Journal of Chemical Physics, 1996, 100(33): 14093-14102. DOI:10.1021/jp9603694 |
| [16] | Zhao X J, Zhao P S, Xu J, et al. Analysis of a winter regional haze event and its formation mechanism in the North China Plain[J]. Atmospheric Chemistry and Physics, 2013, 13(11): 5685-5696. DOI:10.5194/acp-13-5685-2013 |
| [17] | Huang R J, He Y, Duan J, et al. Contrasting sources and processes of particulate species in haze days with low and high relative humidity in wintertime Beijing[J]. Atmospheric Chemistry and Physics, 2020, 20(14): 9101-9114. DOI:10.5194/acp-20-9101-2020 |
| [18] | Han B, Wang Y L, Zhang R, et al. Comparative statistical models for estimating potential roles of relative humidity and temperature on the concentrations of secondary inorganic aerosol: statistical insights on air pollution episodes at Beijing during January 2013[J]. Atmospheric Environment, 2019, 212: 11-21. DOI:10.1016/j.atmosenv.2019.05.025 |
| [19] | Li L, An J Y, Zhou M, et al. Source apportionment of fine particles and its chemical components over the Yangtze River Delta, China during a heavy haze pollution episode[J]. Atmospheric Environment, 2015, 123: 415-429. DOI:10.1016/j.atmosenv.2015.06.051 |
| [20] | Yang Y R, Liu X G, Qu Y, et al. Formation mechanism of continuous extreme haze episodes in the megacity Beijing, China, in January 2013[J]. Atmospheric Research, 2015, 155: 192-203. DOI:10.1016/j.atmosres.2014.11.023 |
| [21] |
谢放尖, 郑新梅, 窦焘焘, 等. 南京地区细颗粒物污染输送影响及潜在源区[J]. 环境科学, 2023, 44(6): 3071-3079. Xie F J, Zheng X M, Dou T T, et al. Transport influence and potential sources of PM2.5 pollution for Nanjing[J]. Environmental Science, 2023, 44(6): 3071-3079. |
| [22] | Yarwood G, Wilson G, Morris R. Development of the CAMx particulate source apportionment technology (PSAT)-final report[R]. Environment International Corporation, 2005. 478-492. |
| [23] | Wagstrom K M, Pandis S N, Yarwood G, et al. Development and application of a computationally efficient particulate matter apportionment algorithm in a three-dimensional chemical transport model[J]. Atmospheric Environment, 2008, 42(22): 5650-5659. DOI:10.1016/j.atmosenv.2008.03.012 |
| [24] | Wu J B, Wang Z F, Wang Q, et al. Development of an on-line source-tagged model for sulfate, nitrate and ammonium: a modeling study for highly polluted periods in Shanghai, China[J]. Environmental Pollution, 2017, 221: 168-179. DOI:10.1016/j.envpol.2016.11.061 |
| [25] | Wang X Q, Wei W, Cheng S Y, et al. Characteristics and classification of PM2.5 pollution episodes in Beijing from 2013 to 2015[J]. Science of the Total Environment, 2018, 612: 170-179. DOI:10.1016/j.scitotenv.2017.08.206 |
| [26] | Lu M M, Tang X, Wang Z F, et al. Investigating the transport mechanism of PM2.5 pollution during January 2014 in Wuhan, Central China[J]. Advances in Atmospheric Sciences, 2019, 36(11): 1217-1234. DOI:10.1007/s00376-019-8260-5 |
| [27] | Wang L T, Wei Z, Wei W, et al. Source apportionment of PM2.5 in top polluted cities in Hebei, China using the CMAQ model[J]. Atmospheric Environment, 2015, 122: 723-736. DOI:10.1016/j.atmosenv.2015.10.041 |
| [28] | Wang Y J, Li L, Chen C H, et al. Source apportionment of fine particulate matter during autumn haze episodes in Shanghai, China[J]. Journal of Geophysical Research: Atmospheres, 2014, 119(4): 1903-1914. DOI:10.1002/2013JD019630 |
| [29] | Shen J Y, Zhao Q B, Cheng Z, et al. Insights into source origins and formation mechanisms of nitrate during winter haze episodes in the Yangtze River Delta[J]. Science of the Total Environment, 2020, 741. DOI:10.1016/j.scitotenv.2020.140187 |
| [30] |
郝新妮, 肖浩, 李亲凯, 等. 天津冬夏季PM2.5中二次无机离子的特征及重污染事件分析——基于连续两年的观测[J]. 环境化学, 2022, 41(10): 3288-3298. Hao X N, Xiao H, Li Q K, et al. Characteristics of secondary inorganic ions in PM2.5 and study of heavy pollution events in winter and summer in Tianjin-based on observations for two consecutive years[J]. Environmental Chemistry, 2022, 41(10): 3288-3298. |
| [31] |
肖致美, 徐虹, 蔡子颖, 等. 2020年天津市两次重污染天气污染特征分析[J]. 环境科学, 2020, 41(9): 3879-3888. Xiao Z M, Xu H, Cai Z Y, et al. Characterization of two heavy pollution episodes in Tianjin in 2020[J]. Environmental Science, 2020, 41(9): 3879-3888. |
| [32] |
元洁, 刘保双, 程渊, 等. 2017年1月天津市区PM2.5化学组分特征及高时间分辨率来源解析研究[J]. 环境科学学报, 2018, 38(3): 1090-1101. Yuan J, Liu B S, Cheng Y, et al. Study on characteristics of PM2.5 and chemical components and source apportionment of high temporal resolution in January 2017 in Tianjin urban area[J]. Acta Scientiae Circumstantiae, 2018, 38(3): 1090-1101. |
| [33] | Zhang W H, Peng X, Bi X H, et al. Source apportionment of PM2.5 using online and offline measurements of chemical components in Tianjin, China[J]. Atmospheric Environment, 2021, 244. DOI:10.1016/j.atmosenv.2020.117942 |
| [34] | Peng X, Liu X X, Shi X R, et al. Source apportionment using receptor model based on aerosol mass spectra and 1 h resolution chemical dataset in Tianjin, China[J]. Atmospheric Environment, 2019, 198: 387-397. DOI:10.1016/j.atmosenv.2018.11.018 |
| [35] |
牛宏宏, 王宝庆, 刘博薇, 等. 天津冬季PM2.5中水溶性无机离子污染特征研究[J]. 环境污染与防治, 2019, 41(5): 592-595. Niu H H, Wang B Q, Liu B W, et al. Characteristics of water-soluble inorganic ions in PM2.5 during winter in Tianjin[J]. Environmental Pollution & Control, 2019, 41(5): 592-595. |
| [36] |
蔡子颖, 杨旭, 韩素芹, 等. 基于天气背景天津大气污染输送特征分析[J]. 环境科学, 2020, 41(11): 4855-4863. Cai Z Y, Yang X, Han S Q, et al. Transport characteristics of air pollution in Tianjin based on weather background[J]. Environmental Science, 2020, 41(11): 4855-4863. |
| [37] | Hao T Y, Cai Z Y, Chen S C, et al. Transport pathways and potential source regions of PM2.5 on the west coast of Bohai Bay during 2009-2018[J]. Atmosphere, 2019, 10(6). DOI:10.3390/atmos10060345 |
| [38] |
王自发, 谢付莹, 王喜全, 等. 嵌套网格空气质量预报模式系统的发展与应用[J]. 大气科学, 2006, 30(5): 778-790. Wang Z F, Xie F Y, Wang X Q, et al. Development and application of nested air quality prediction modeling system[J]. Chinese Journal of Atmospheric Sciences, 2006, 30(5): 778-790. |
| [39] |
马琳, 魏巍, 张稳定, 等. 2016年秋季新乡市空气质量模式预报效果评估[J]. 中国环境监测, 2017, 33(5): 89-94. Ma L, Wei W, Zhang W D, et al. Evaluation on air quality forecasting model in the fall of 2016 in Xinxiang City, Henan Province[J]. Environmental Monitoring in China, 2017, 33(5): 89-94. |
| [40] | Walmsley J L, Wesely M L. Modification of coded parametrizations of surface resistances to gaseous dry deposition[J]. Atmospheric Environment, 1996, 30(7): 1181-1188. DOI:10.1016/1352-2310(95)00403-3 |
| [41] | Chang J S, Brost R A, Isaksen I S A, et al. A three-dimensional Eulerian acid deposition model: physical concepts and formation[J]. Journal of Geophysical Research: Atmospheres, 1987, 92(D12): 14681-14700. DOI:10.1029/JD092iD12p14681 |
| [42] | Zaveri R A, Peters L K. A new lumped structure photochemical mechanism for large-scale applications[J]. Journal of Geophysical Research: Atmospheres, 1999, 104(D23): 30387-30415. DOI:10.1029/1999JD900876 |
| [43] | Nenes A, Pandis S N, Pilinis C. ISORROPIA: a new thermodynamic equilibrium model for multiphase multicomponent inorganic aerosols[J]. Aquatic Geochemistry, 1998, 4(1): 123-152. DOI:10.1023/A:1009604003981 |
| [44] | Li J, Wang Z, Zhuang G, et al. Mixing of Asian mineral dust with anthropogenic pollutants over East Asia: a model case study of a super-duststorm in March 2010[J]. Atmospheric Chemistry and Physics, 2012, 12(16): 7591-7607. DOI:10.5194/acp-12-7591-2012 |
| [45] | Skamarock W C, Klemp J B, Dudhia J, et al. A description of the advanced research WRF version 3 [R]. NCAR Technical Note NCAR/TN-475+STR, 2008, Mesoscale and Microscale Meteorology Division. National Center for Atmospheric Research. Boulder, 475. |
| [46] | Mlawer E J, Taubman S J, Brown P D, et al. Radiative transfer for inhomogeneous atmospheres: RRTM, a validated correlated-k model for the longwave[J]. Journal of Geophysical Research: Atmospheres, 1997, 102(D14): 16663-16682. DOI:10.1029/97JD00237 |
| [47] | Chou M D, Suarez M J. An efficient thermal infrared radiation parameterization for use in general circulation models[M]. Greenbelt: National Aeronautics and Space Administration, Goddard Space Flight Center, 1994. |
| [48] | Ek M B, Mitchell K E, Lin Y, et al. Implementation of Noah land surface model advances in the national centers for environmental prediction operational mesoscale Eta model[J]. Journal of Geophysical Research: Atmospheres, 2003, 108(D22). DOI:10.1029/2002JD003296 |
| [49] | Grell G A, Peckham S E, Schmitz R, et al. Fully coupled "online" chemistry within the WRF model[J]. Atmospheric Environment, 2005, 39(37): 6957-6976. DOI:10.1016/j.atmosenv.2005.04.027 |
| [50] | Janjić Z I. The step-mountain Eta coordinate model: further developments of the convection, viscous sublayer, and turbulence closure schemes[J]. Monthly Weather Review, 1994, 122(5): 927-945. DOI:10.1175/1520-0493(1994)122<0927:TSMECM>2.0.CO;2 |
| [51] | Lin Y L, Farley R D, Orville H D. Bulk parameterization of the snow field in a cloud model[J]. Journal of Applied Meteorology and Climatology, 1983, 22(6): 1065-1092. DOI:10.1175/1520-0450(1983)022<1065:BPOTSF>2.0.CO;2 |
| [52] | Brasseur G P, Hauglustaine D A, Walters S, et al. MOZART, a global chemical transport model for ozone and related chemical tracers: 1. Model description[J]. Journal of Geophysical Research: Atmospheres, 1998, 103(D21): 28265-28289. DOI:10.1029/98JD02397 |
| [53] | Hauglustaine D A, Brasseur G P, Walters S, et al. MOZART, a global chemical transport model for ozone and related chemical tracers: 2. Model results and evaluation[J]. Journal of Geophysical Research: Atmospheres, 1998, 103(D21): 28291-28335. DOI:10.1029/98JD02398 |
| [54] | Janssens-Maenhout G, Crippa M, Guizzardi D, et al. HTAP_v2.2: a mosaic of regional and global emission grid maps for 2008 and 2010 to study hemispheric transport of air pollution[J]. Atmospheric Chemistry and Physics, 2015, 15(19): 11411-11432. DOI:10.5194/acp-15-11411-2015 |
| [55] | Randerson J T, Van Der Werf G R, Giglio L, et al. Global fire emissions database, version 4.1 (GFEDv4)[R]. Oak Ridge: ORNL Distributed Active Archive Center, 2017. |
| [56] | Van Der Werf G R, Randerson J T, Giglio L, et al. Global fire emissions and the contribution of deforestation, savanna, forest, agricultural, and peat fires (1997-2009)[J]. Atmospheric Chemistry and Physics, 2010, 10(23): 11707-11735. DOI:10.5194/acp-10-11707-2010 |
| [57] | Sindelarova K, Granier C, Bouarar I, et al. Global data set of biogenic VOC emissions calculated by the MEGAN model over the last 30 years[J]. Atmospheric Chemistry and Physics, 2014, 14(17): 9317-9341. DOI:10.5194/acp-14-9317-2014 |
| [58] | Li J, Wang Z B, Akimoto H, et al. Near-ground ozone source attributions and outflow in central eastern China during MTX2006[J]. Atmospheric Chemistry and Physics, 2008, 8(24): 7335-7351. DOI:10.5194/acp-8-7335-2008 |
| [59] |
冯晓青. 代煤工程前后天津市冬季PM2.5污染特征与来源对比研究[D]. 天津: 天津大学, 2019. Feng X Q. In sight of pollution characteristics and sources comparison of PM2.5 before and after the coal replacing project in Tianjin wintertime[D]. Tianjin: Tianjin University, 2019. |
| [60] | Quan J N, Tie X, Zhang Q, et al. Characteristics of heavy aerosol pollution during the 2012-2013 winter in Beijing, China[J]. Atmospheric Environment, 2014, 88: 83-89. DOI:10.1016/j.atmosenv.2014.01.058 |
| [61] | Ianniello A, Spataro F, Esposito G, et al. Occurrence of gas phase ammonia in the area of Beijing (China)[J]. Atmospheric Chemistry and Physics, 2010, 10(19): 9487-9503. DOI:10.5194/acp-10-9487-2010 |
| [62] | Petetin H, Sciare J, Bressi M, et al. Assessing the ammonium nitrate formation regime in the Paris megacity and its representation in the CHIMERE model[J]. Atmospheric Chemistry and Physics, 2016, 16(16): 10419-10440. DOI:10.5194/acp-16-10419-2016 |
| [63] | Wang G H, Zhang R Y, Gomez M E, et al. Persistent sulfate formation from London fog to Chinese haze[J]. Proceedings of the National Academy of Sciences of the United States of America, 2016, 113(48): 13630-13635. |
| [64] | Elshorbany Y F, Kurtenbach R, Wiesen P, et al. Oxidation capacity of the city air of Santiago, Chile[J]. Atmospheric Chemistry and Physics, 2009, 9(6): 2257-2273. DOI:10.5194/acp-9-2257-2009 |
| [65] | Sun Y L, Zhuang G S, Tang A H, et al. Chemical characteristics of PM2.5 and PM10 in haze-fog episodes in Beijing[J]. Environmental Science & Technology, 2006, 40(10): 3148-3155. |
| [66] | Wang Y, Zhuang G S, Zhang X Y, et al. The ion chemistry, seasonal cycle, and sources of PM2.5 and TSP aerosol in Shanghai[J]. Atmospheric Environment, 2006, 40(16): 2935-2952. DOI:10.1016/j.atmosenv.2005.12.051 |
| [67] | Lin M Y, Oki T, Bengtsson M, et al. Long-range transport of acidifying substances in east Asia-part Ⅱ: source-receptor relationships[J]. Atmospheric Environment, 2008, 42(24): 5956-5967. DOI:10.1016/j.atmosenv.2008.03.039 |
| [68] | Lu M M, Tang X, Feng Y C, et al. Nonlinear response of SIA to emission changes and chemical processes over eastern and central China during a heavy haze month[J]. Science of the Total Environment, 2021, 788. DOI:10.1016/j.scitotenv.2021.147747 |
| [69] | Chen D, Liu Z Q, Fast J, et al. Simulations of sulfate-nitrate-ammonium (SNA) aerosols during the extreme haze events over northern China in October 2014[J]. Atmospheric Chemistry and Physics, 2016, 16(16): 10707-10724. DOI:10.5194/acp-16-10707-2016 |