 2023, Vol. 44
2023, Vol. 44



2. 西南林业大学云南滇池湿地生态系统国家定位观测研究站, 昆明 650224;
3. 中国科学院城市环境研究所城市环境与健康重点实验室, 厦门 361021
2. National Wetland Ecosystem Fixed Research Station of Yunnan Dianchi, Southwest Forestry University, Kunming 650224, China;
3. Key Laboratory of Urban Environment and Health, Institute of Urban Environment, Chinese Academy of Sciences, Xiamen 361021, China
近年来, 抗生素在医疗、畜牧养殖和农业生产中的广泛使用导致环境中的细菌在抗生素的选择压力下产生耐药性[1], 并形成耐药菌[2]. 抗生素耐药性已经成为全球公共卫生关注的焦点, 一旦微生物对抗生素产生耐药性, 临床上一些常见的感染性疾病将面临无药可医的境地.鉴于这一现象的普遍性, 人们将越来越多的注意力转向可能有助于抗生素抗性基因(antibiotic resistant genes, ARGs)在临床领域之外传播的环境途径[3].过去数十年的研究证明, 随着人类活动对环境影响强度的增加, ARGs污染的程度也随之增加[4], 人类活动与ARGs环境污染之间正相关的关系也不断被证实[5].因此, 识别抗性基因可能的污染来源并了解人类活动与ARGs产生的关系就显得尤为重要.此外, 由于抗生素耐药性是一种自然现象, 某些类型的抗性基因是环境中天然存在的[6~8], 因此对比研究自然产生的抗性基因与人为污染产生的抗性基因(包括类型、含量、传播和风险等)对抗性基因的溯源跟踪以及污染控制至关重要.
目前关于水环境中抗性基因的污染研究多集中于经济发达和人口稠密地区[9], 而对于经济发展相对落后, 人口密度较小的高原湿地的研究则相对较少.湿地被称为“地球之肾”, 发挥着蓄水保土、净化水质、调节气候和维护生物多样性等重要的生态功能.其中, 高原湿地面积占据全国湿地资源的一半, 高原湿地独特的地理环境及较高的海拔使其较少受人类活动的干扰, 是相对原始的生境[10].然而, 近年来, 随着高原人类活动的加剧, 高原湿地呈现变异敏感度高、稳定性差等一系列生态脆弱性特征, 是典型的生态脆弱带和敏感地区[11], 人类活动对高原湿地中ARGs赋存和传播的影响备受关注[12, 13].高原湿地中的河流湿地在为生态系统和人类社会提供重要资源的同时也是各种生物污染物的蓄水池[14], 这些生物污染物不可避免地从不同的污水来源汇集, 可引起微生物群落发生变化, 甚至导致ARGs向病原体传播[15], 降低对病原体的治疗潜力, 引发人类健康高风险[16].为了准确识别由自然以外的人类活动导致的ARGs污染, 首先必须排除本土环境中原生ARGs的干扰.
本文选取云贵高原的鹤庆草海国家湿地公园为研究区, 该区域因地壳在差异抬升过程中断裂形成诸多泉眼, 这些泉眼发源自地下水, 水质天然洁净, 汇集成河流的源头, 是重要的饮用水源;然而在河流下游, 因流经村庄, 大量居民生活污水在下游汇集, 该段河流不可避免地成为各种污水来源的蓄积池.为对比研究自然生境及人类活动干扰下的ARGs的赋存与传播, 沿河流梯度分别等距采集上游水源地(泉眼)和下游(受污水排放影响的纳污河流段)河流中的沉积物样品, 对河流中的抗生素及四环素类抗性基因进行检测, 并对其赋存和分布特征与环境因子、微生物群落和病原菌进行分析, 以期为高原湿地生态系统中ARGs的溯源跟踪和污染控制提供基础数据.
1 材料与方法 1.1 样品采集研究区位于云南省大理市鹤庆草海国家湿地公园, 以距离村庄远近将河流湿地划分为上游(upstream, US)和下游(downstream, DS), 上、下游相距250 m.上游位于国家湿地公园内, 平均海拔为2 206 m, 该段河流发源于凤凰山脚下的一处泉眼, 受国家和政府严格保护, 且距离村庄距离较远, 水质天然洁净无污染, 排除了养殖、放牧和污水排放等直接人类活动的干扰, 可看作是原始生境.下游位于排污口下方, 平均海拔为2 199 m, 主要污染源来自当地居民生活污水排放, 为主要的纳污河流段.利用土壤取样器沿河流梯度分别于上游(间距为3 m)和下游(间距为10 m)各等距采集3个沉积物样本, 每个样品由5个质量为100 g的子样本均匀混合而成.上游样品标记为US1、US2和US3, 下游样品标记为DS1、DS2和DS3(图 1).样品剔除大块砂砾和生物残体后装于密封袋中用于基础理化指标的测定, 同时取约5 g样品装入5 mL无菌离心管中, 封口后立即放入盛有冰袋的采样箱中, 用液氮保存运至实验室预处理.将离心管中的样品储存于-80℃冰箱中, 用于分子生物学实验及沉积物中微生物DNA提取.

|
图 1 采样点示意 Fig. 1 Sampling sites |
共进行9项基本理化指标和16种抗生素的测定.土壤含水率(SWC)采用烘干称重法;pH值采用酸度计电位法;土壤有效磷(Olsen-P)采用钼锑抗比色法;土壤总有机碳(TOC)采用总有机碳分析仪(vario TOC, 德国)测定;土壤总氮(TN)和总磷(TP)用浓硫酸消煮后用连续流动分析仪(AA3, Bran+LuebbeCrop, 德国)测定;土壤硝态氮(NO3--N)和铵态氮(NH4+-N)用氯化钾浸提法, 然后使用连续流动分析仪(德国SEAL Analytical AA3)测定;阳离子交换量(CEC)采用三氯化六氨合钴浸提-分光光度法.参照之前使用的研究方法[17], 使用HPLC-MS/MS系统(LCMS-8040系统, 日本岛津)对4类共16种抗生素进行定量测定, 包括四环素类:四环素(TC)、土霉素(OTC)、金霉素(CTC)和强力霉素(DOC);磺胺类:磺胺嘧啶(SDZ)、磺胺甲噁唑(SMX)、磺胺二甲嘧啶(SMZ)、磺胺间甲氧嘧啶(SMM)、磺胺喹恶啉(SCX)、磺胺地索辛(SCZ)和磺胺氯吡嗪(SDM);喹诺酮类:诺氟沙星(NFC)、氧氟沙星(OFC)、环丙沙星(CFC)和恩诺沙星(EFC);大环内酯类:罗红霉素(RTM).
1.3 DNA提取及PCR定量根据制造商的说明, 使用MoBio PowerSoil DNA提取试剂盒(MoBio Carlsbad, CA, USA)提取土壤DNA, 然后使用1%的琼脂糖凝胶检测DNA完整性以确定质量, 提取后的DNA样品于-80℃下保存备用.采用SYBR Green法对土样中的四环素类抗生素抗性基因进行检测, 共使用26条引物[18]扩增沉积物DNA中的靶基因, 包括四环素类抗生素的抗性基因(25条引物)和16S rRNA基因, 每一套引物都包括一个非模板阴性对照.PCR反应条件为:95℃下热变性10 min, 随后进行40个循环, 每个循环包括95℃下变性30 s, 60℃退火温度下反应30 s, 72℃下延伸10 s, 自动生成扩增曲线, 后冷却至40℃, 并维持30 s.反应结束后获得Ct值(目标扩增产物达到设定阈值的扩增循环数).所有的PCR定量都进行3次技术重复, 而只有在3个技术重复中均被检出的基因, 才将该基因判定为阳性.先计算扩增的基因数, 然后根据下式计算每个扩增基因的相对基因拷贝数.
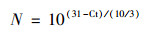
|
式中, Ct为PCR定量结果, N为相对基因拷贝数, 31为检测限.然后通过归一化16S rRNA基因拷贝数, 将每个基因的相对拷贝数转化为绝对拷贝数(copies ·g-1).在定量16S rRNA基因拷贝数时, 需考虑DNA提取效率和可能的PCR反应抑制, 采用适当的稀释步骤.
1.4 细菌群落分析利用引物338F(5′-ACTCCTACGGGAGGCAGC AG-3′)和806R(5′-GGACTACHVGGGTWTCTAAT-3′)对16S rRNA基因V3~V4可变区进行PCR扩增, 使用NEXTFLEXRapid DNA-Seq Kit进行建库, 用Illumina-MiSeq高通量测序平台进行测序.MiSeq测序得到的PE reads首先根据overlap关系进行拼接, 同时对序列质量进行质控和过滤, 然后采用RDP classifier贝叶斯算法以97%相似水平对OTU进行聚类分析, 细菌和病原菌分别基于Silva数据库、Human_HPB数据库进行比对注释, 而后进行相应的物种分类学分析.
1.5 数据分析用Origin绘制抗生素含量分布图, Student t检验(Student's t test)对基础理化指标进行差异性检验.为明确抗性基因的检出及丰度, 分别用R语言、Origin绘制韦恩图和柱形图;采用R语言中的Vegan包和ggplot2包进行主坐标轴分析(principal coordinates analysis, PCoA), 以考察抗性基因的分布差异. 细菌16S rRNA原始测序数据基于QIIME(Version 1.7.0)进行处理, 在进行微生物分析之前, reads首先被归一化到等序深度;通过香农指数、辛普森指数、艾斯指数、赵氏指数和覆盖度来计算微生物的α多样性;同时以河流梯度为约束条件, 基于Bray-Curtis距离, 使用R语言vegan包中的Capscale函数对微生物β多样性进行CAP分析(canonical analysis of principal coordinates), 此处使用约束排序的目的是为了评估河流污染梯度对微生物群落的约束效应的重要性.为进一步考察AGRs与环境因子、微生物之间的关系, 使用R语言中的pleatmap和ggplot2包绘制理化指标、抗生素含量和ARGs的相关性热图, Psych包计算ARGs与微生物之间的Spearman相关性系数, 基于相关性数据采用Gephi软件进行网络可视化.以上所有统计分析及绘图均在SPSS 25.0、Origin 2018、Gephi和R语言中完成.
2 结果与分析 2.1 理化指标及抗生素含量分布特征由表 1可知, 下游与上游相比, pH和铵态氮含量呈下降趋势, 但差异不显著, 而含水率、总有机碳、总氮、总磷、硝态氮、有效磷和阳离子交换量均呈上升趋势, 其中, 总氮和硝态氮在上、下游差异极显著(P < 0.01), 总氮在下游较上游增加了2.16倍, 硝态氮增加了12.26倍;总有机碳在上、下游差异显著(P < 0.05), 在下游较上游增加了2.81倍.
|
|
表 1 上、下游沉积物基本理化指标及差异比较1) Table 1 Comparison of soil physical and chemical indexes between upstream and downstream |
上、下游沉积物中共检出4类16种抗生素(图 2), 包括4环素类(TC、OTC、CTC、DOC)、磺胺类(SDZ、SMX、SMZ、SMM、SCX、SDM、SCZ)、喹诺酮类(NFC、OFC、CFC、EFC)和大环内酯类(RTM).上、下游抗生素含量差异明显, 上游抗生素总含量为195.01 μg ·kg-1, 下游抗生素总含量为1 415.89 μg ·kg-1, 下游沉积物中的抗生素含量约为上游的7倍, 远高于上游.从检出率来看, 磺胺类抗生素检出率为73.8%, 四环素类抗生素检出率为71%, 两类抗生素是沉积物中检出最为频繁的抗生素种类.四环素类抗生素总含量为103.65~2 185 μg ·kg-1, 磺胺类抗生素总含量为17.45~105.12 μg ·kg-1, 喹诺酮类抗生素总含量为0~31.25 μg ·kg-1, 大环内酯类抗生素总含量为0~0.85 μg ·kg-1, 四环素类抗生素是检出含量最高的抗生素种类.此外, 四环素类抗生素在上、下游含量差异最明显, 上游的含量为127.80 μg ·kg-1, 下游的含量为1 354.67 μg ·kg-1, 下游约为上游的10倍.从显著性检验结果来看, 除OTC、CTC、DOC、EFC和RTM这5种抗生素在上、下游差异显著(P < 0.05)外, 其余均不显著.

|
相同小写字母表示无显著差异, 不同小写字母表示在0.05水平下差异性显著 图 2 抗生素在上、下游沉积物含量 Fig. 2 Contents of antibiotics in upstream and downstream sediments |
由图 3(a)可知, 上、下游沉积物中共检出15种四环素类ARGs.上游检出7种ARGs, 分别是tetB、tetbP、tetD、tetG、tetL、tetPA和tetPB;下游检出13种ARGs, 分别是tet(36)、tet(38)、tetbP、tetD、tetE、tetG、tetM、tetO、tetPA、tetPB、tetQ、tetW和tetX, 其中, tetPA、tetD、tetG、tetPB和tetbP这5种抗性基因在上、下游均有检出.由图 3(b)可知, 上游四环素类ARGs绝对丰度为1.20×108 copies ·g-1, 检出率为46.67%;下游四环素类ARGs绝对丰度为7.94×107 copies ·g-1, 检出率为86.67%.其中丰度最高的抗性基因是tetPA, 占所有检出抗性基因绝对丰度的73.5%, 其在上、下游的丰度均为最高, 且上游高于下游, 占上游检出基因丰度的95.83%, 占下游检出基因丰度的39.29%, 是引起下游基因总丰度低于上游的关键基因; 除此之外, 其余抗性基因的丰度在下游较上游均明显增加.对上、下游共同检出的5种抗性基因进行差异性检验, 结果均无显著性差异(P>0.05), 而下游新增的8种基因占下游总丰度的43.44%.对上、下游沉积物中的抗性基因进行主成分分析[图 3(c)], 结果表明:PC1(主成分1)可以解释ARGs丰度变异的56.72%, PC2(主成分2)可以解释ARGs丰度变异的25.75%, PC1和PC2两轴共同解释了82.47%.抗性基因自上游至下游依次沿PC1轴分布, 上游与下游的抗性基因组成有明显的差异性, 且上游各个样品的抗性基因组成较下游更为相似.

|
(a)四环素类ARGs在上、下游沉积物中的分布;(b)四环素类ARGs在上、下游沉积物中的绝对丰度;(c)四环素类ARGs在上、下游沉积物中的主成分分析; 1.tetB, 2.tetL, 3.tetPA, 4.tetD, 5.tetPB, 6.tetbP, 7.tetG, 8.tet(36), 9.tet(38), 10.tetE, 11.tetM, 12.tetO, 13.tetQ, 14.tetW, 15.tetX; 相同小写字母表示无显著差异, 不同小写字母表示在0.05水平下差异性显著 图 3 上、下游沉积物中ARGs的分布 Fig. 3 Distribution of ARGs in upstream and downstream sediments |
由表 2可知, 在97%相似水平上, Coverage指数均在98%以上, 说明本次测序结果可充分反映微生物的真实情况.从各个多样性指数的差异性比较结果来看, 香农指数、辛普森指数、艾斯指数和赵氏指数在上、下游均无显著性差异.
|
|
表 2 上、下游沉积物中细菌丰富度和多样性指数 Table 2 Index table of bacterial abundance and diversity in upstream and downstream sediments |
上、下游沉积物样品共获得了361 068条高质量的16S rRNA基因序列, 平均每个样本有60 178条序列, 按照97%的相似性对非重复序列(不含单序列)进行OTU聚类, 在聚类过程中去除嵌合体, 得到8 529个OTU代表序列.首先基于OTU进行CAP分析, 结果表明样点自上游至下游依次沿CAP1轴分布(图 4), 上、下游微生物群落组成差异显著(P < 0.05).上游样点之间的距离较远, 微生物群落组成较为分散, β多样性差异较大;下游样点之间的距离较小, 微生物群落组成较为集中, β多样性差异较小.说明随着污染源的输入, 河流下游的微生物群落组成越来越相似.

|
采样梯度以箭头方向指示, 箭头长短表示各个采样点之间微生物群落组成的差异大小 图 4 采样梯度约束下的细菌群落在OTU水平上的排序 Fig. 4 OTU-based bacterial community difference distribution between upstream and downstream using canonical analysis of principal coordinates |
对沉积物中的细菌进行注释, 8 529个可操作分类单元(OTUs)分属: 64门、189纲、436目、686科1 178属和2 615种.在门水平上[图 5(a)], 丰度排名前10的细菌门类分别是: 变形菌门(Proteobacteria)、酸杆菌门(Acidobacteriota)、绿弯菌门(Chloroflexi)、拟杆菌门(Bacteroidota)、Actinobacteriota、Desulfobacterota、粘菌门(Myxococcota)、Gemmatimonadota、Methylomirabilota和厚壁菌门(Firmicutes).上、下游的沉积物细菌组成在门水平上具有一定的差异性, 在丰度排名前10的门类群中, 酸杆菌门(Acidobacteriota)和Desulfobacterota的相对丰度在上游中高于下游, 其余均低于下游.在科水平上[图 5(b)], 丰度排名前10的科分别是: 伯克氏菌科(Burkholderiaceae)、鞘脂单胞菌科(Sphingomonadaceae)、厌氧绳菌科(Anaerolineaceae)、Anaeromyxobacteraceae、芽单胞菌科(Gemmatimonadaceae)、丛毛单胞菌科(Comamonadaceae)、SC-I-84、Vicinamibacteraceae、亚硝化单胞菌科(Nitrosomonadaceae)和Methylomirabilaceae.上、下游的沉积物细菌组成在科水平上具有一定的差异性, 其中, 厌氧绳菌科(Anaerolineaceae)、SC-I-84和Methylomirabilaceae在上游的相对丰度高于下游, 其余均低于下游.基于HPB数据库进行比对注释, 上、下游共检出27种病原菌[图 5(c)], 总丰度为25.21%, 其中上游检出23种病原菌, 丰度为12.05%;下游检出25种病原菌, 丰度为13.16%, 下游较上游病原菌种类和数量均增加.这些病原菌分属于放线菌门(Actinobacteria)、拟杆菌门(Bacteroidetes)、厚壁菌门(Firmicutes)、变形菌门(Proteobacteria)和衣原体门(Chlamydiae)这5个菌门, 巴东氏菌(Bartonella henselae)和溃疡分枝杆菌(Mycobacterium ulcerans)的丰度分别为13.32%和6.45%, 为主要的优势病原菌.

|
(a)丰度排名前15的细菌在门水平上的群落组成;(b)丰度排名前15的细菌在科水平上的群落组成;(c) 病原菌在上、下游沉积物中的丰度 图 5 上、下游沉积物中的细菌及病原菌群落组成 Fig. 5 Composition of bacterial and pathogenic bacteria communities in upstream and downstream sediments |
4种四环素类抗生素和9项理化指标与ARGs的相关性热图如图 6所示. tet(36)与OTC呈极显著正相关(P < 0.01), tetW与OTC呈显著正相关(P < 0.05);tetM、tetG与CTC呈极显著正相关(P < 0.001);tetD与DOC呈显著正相关(P < 0.05), tet(36)、tetW与DOC呈极显著正相关, 其显著性结果分别为P < 0.01和P < 0.001;tetPA与TC呈显著负相关(P < 0.05), tetD、tet(36)与TC呈显著正相关(P < 0.05), tetW与之呈极显著正相关(P < 0.01).以上结果表明, 四环素类抗生素含量的积累会诱导四环素类ARGs的扩散和转移.
就理化指标来看, pH、含水率、总氮、铵态氮和阳离子交换量均与四环素类ARGs无显著相关性, 有效磷、总有机碳、硝态氮和总磷与四环素类ARGs存在一定程度的相关性.有效磷与tetPB呈显著负相关(P < 0.05);总有机碳与tetM和tetG呈极显著正相关(P < 0.01), 与tet(36)呈显著正相关(P < 0.05);硝态氮与tetM和tetG均呈极显著正相关(P < 0.001);总磷与tetO、tetX、tetQ和tet(38)均呈显著正相关(P < 0.05).

|
1.tetPA, 2.tetO, 3.tetE, 4.tetX, 5.tetQ, 6.tetM, 7.tetD, 8.tetG, 9.tet(38), 10.tet(36), 11.tetW, 12.tetPB, 13.tetbP, 14.tetB, 15.tetL; *表示P < 0.05, **表示P < 0.01, ***表示P < 0.001 图 6 抗生素含量、理化指标和ARGs相关性 Fig. 6 Correlation between antibiotic content, physical, and chemical indexes and ARGs |
为探究微生物群落结构和ARGs之间的关系, 基于微生物相对丰度(属水平)和四环素类ARGs相对丰度计算ARGs和微生物之间所有可能成对的Spearman相关系数来构建相关性矩阵, 探索ARGs和微生物之间的共现性, 并进行可视化[图 7(a)], 详细网络图参数见表 3. tet(36)、tetQ、tetX、tet(38)和tetW是连接度较高的ARGs, 其分别与60、51、51、49和49个菌属呈显著正相关, 表明这些抗性基因可能有多个潜在宿主;变形菌门、厚壁菌门和拟杆菌门是连接度较高的微生物菌门, 其中变形菌门的相对丰度为35.6%、厚壁菌门的相对丰度为15.1%、拟杆菌门的相对丰度为13.2%, 这些菌门可能是多个ARGs的潜在宿主.图 7(c)为四环素类ARGs及其对应的相关性的微生物丰度, 其展示了不同四环素类ARGs与不同的微生物(门水平)具有不同的相关性.

|
(a)四环素类抗性基因与细菌群落(属)相关性分析(r>0.8, P < 0.05), (b)四环素类抗性基因与病原菌相关性分析(r>0.8, P < 0.05), (c)与四环素类抗性基因显著相关的细菌属在门水平上的分布, (d)与四环素类抗性基因显著相关的病原菌在门水平上的分布;c1.tet(36), c2.tetQ, c3.tetX, c4.tet(38), c5.tetW, c6.tetD, c7.tetL, c8.tetM, c9.tetPB, c10.tetE, c11.tetO, c12.tetbP, c13. tetG, c14.tetB, c15.tetPA;d1.tet(38), d2.tetQ, d3.tetO, d4.tetPB, d5.tetX, d6.tet(36), d7.tetE, d8.tetL, d9.tetB, d10.tetbP, d11.tetD, d12.tetG, d13.tetM, d14.tetW 图 7 微生物群落结构和ARGs的关系 Fig. 7 Relationship between microbial community structure and ARGs |
|
|
表 3 网络图参数 Table 3 Network diagram parameters |
为进一步明确病原菌与四环素类ARGs间的内在联系, 将病原菌(种水平)的相对丰度和四环素类ARGs的相对丰度进行网络分析, 探索ARGs和病原菌之间的共现性, 并进行可视化[图 7(b)], 相关网络图参数见表 3.在检出的27种病原菌中有15种与ARGs呈显著相关(Spearman相关系数r>0.8;P < 0.05), 其中, 巴尔通体氏(Bartonella henselae)与tetO、tetQ和tet(38)呈显著负相关, 砂眼批衣菌(Chlamydia trachomatis)与tet(36)呈显著负相关, 皮面葡萄球菌(Staphylococcus epidermidis)与tetbP呈显著负相关, 其余均与四环素类ARGs呈显著正相关.连接度较高的病原菌有普通拟杆菌(Bacteroides vulgatus)、粪肠球菌(Enterococcus faecalis) 和李斯特菌(Listeria monocytogenes), 说明这些病原菌可能是多种四环素类ARGs的潜在病原菌宿主.下游新增加的tet(38)、tetO和tetQ等8个基因均与病原菌有不同程度的相关性, 在网络图33个相关关系中有23个均与下游新增的基因有关, 占据网络连接度的70%, 说明这些四环素类ARGs可能有多个潜在的微生物宿主.图 7(d)表明不同四环素类ARGs与不同的病原菌(门水平)具有不同的相关性, 与四环素类ARGs相关的病原菌主要分属于Bacteroidetes、Chlamydiae、Firmicutes和Proteobacteria这4个菌门, 其中Firmicutes和Proteobacteria是相关性最高的两个菌门.
3 讨论 3.1 四环素类ARGs分布特征分析四环素类抗生素因使用量大、较为稳定的化学性质以及较强的自我扩增能力, 在环境中广泛分布[19, 20], 目前在地表水、土壤和地下水等环境中均有检出[21].本研究中, 四环素类抗生素是检出含量最高的抗生素种类, 与检出的其他3类抗生素相比, 四环素类抗生素结构复杂, 极易吸附在沉积物中, 从而增加了降解的难度[22].抗生素含量的积累必然会诱导微生物产生相应的抗性基因, 抗性基因是一种新型生物污染物[23], 对环境的危害远比抗生素本身还要大[24].基于此, 本文针对25种目标四环素类ARGs进行检测, 结果有15种四环素类ARGs被检出, 相较于上游, 下游检出的ARGs种类更为丰富.Chen等[25]采用宏基因组方法比较分析南海深层海床和华南珠江口沉积物中抗生素耐药基因的分布特征, 发现被污染的珠江口沉积物中抗性基因在基因型和耐药性机制方面都比深海中的多样性更高, 说明ARGs自然起源于原始环境, 而人类活动加速了ARGs的传播[26], 使微生物能够耐受选择性环境应激, 以应对人类活动的影响.特别地, 本研究中丰度最高的抗性基因为tetPA, 其在上、下游的含量均为最高, 占检出抗性基因总丰度的73.5%, 除此之外, 其余共同检出的抗性基因均为下游高于上游, tetPA是造成上游绝对丰度高于下游的主要优势基因, 推测可能是tetPA这种抗性基因在某些微生物中天然存在.当前已经在自然原生环境中发现了具有临床重要性的ARGs[27], 在自然土壤[28]甚至3万年前的永久冻土沉积物中[7], 已经发现了编码β-内酰胺、四环素和糖肽抗生素抗性的基因;在一项跨地理梯度的污染区域研究中发现背景环境中存在一个本底tet基因库, 同时维持这些库的生态压力并不完全来源于人类活动[29], 表明原始生境中存在的ARGs是自然进化的产物, 在不同环境选择压力下呈现不同程度的富集.
3.2 抗生素含量及理化指标对四环素类ARGs的影响细菌是土壤生态系统的重要组成成分, 对生态环境健康有重要的指示作用.抗生素进入土壤后, 通常会对土壤微生态系统产生破坏, 抑制土壤微生物的生长与繁殖[30].四环素类抗生素对微生物的生态毒性体现在:四环素类抗生素通过同细菌核糖体30S亚基A位点结合, 从而阻止酰胺tRNA与该位点的结合, 抑制细菌翻译过程进而影响菌体蛋白质的合成, 进而抑制细菌的生长繁殖[31].环境中外源抗生素的输入会直接诱导、加速环境中抗生素抗性基因的产生和传播[32], 增加土壤中抗生素抗性基因亚型.本研究的相关性分析表明, 沉积物中抗性基因的含量和丰度与对应的抗生素含量之间存在正相关关系.尤新新[33]在垃圾填埋场渗滤液中发现, 四环素类抗生素(OTC、TC和DC)与四环素类ARGs呈显著相关关系;Wu等[34]研究填埋场滤液中的抗性基因, 发现四环素类抗生素与抗性基因tetQ和tetW存在相关性;Xu等[35]研究污水处理厂及其周围水环境, 发现四环素类抗生素与抗性基因tetW相关;赵赛[36]研究长江口滨岸沉积物, 发现土霉素(OTC)和强力霉素(DXC)与对应的四环素类ARGs显著相关.以上研究说明, 四环素类抗生素的输入会引起四环素类ARGs丰度的增加.
除了抗生素的诱导会对ARGs产生影响外, 环境因子(碳、氮、磷、营养物和电子受体)对ARGs的分布也起到了重要作用[37].本研究中抗性基因的分布与pH、含水率、总氮、铵态氮和阳离子交换量均无显著相关性, 与有效磷、总有机碳、硝态氮和总磷存在一定程度的相关性. Di Cesare等[38]研究表明, tetA、tetB的赋存和分布与微生物总量、总磷和总氮等有关;McKinney等[39]研究也发现废水中四环素类ARGs与总氮、硝态氮和铵态氮有显著相关性.这些环境因子与ARGs的正相关性, 表明碳、氮、磷等营养元素的浓度越高, 越有利于环境微生物的生长, 从而促进ARGs的繁殖与扩增[40].综上所述, 沉积物中四环素类ARGs的浓度分布与污水排放有关, 且污染源中携带的四环素类抗生素是沉积物中ARGs的重要来源, 同时水体中的碳、氮、磷元素能够促进微生物的繁殖, 间接促进ARGs的传播, 因此需要对污染源向河流的排放加以控制与治理, 以降低河流水体中微生物对ARGs的传播和扩散作用.
3.3 微生物群落结构对ARGs分布的影响微生物群落结构是影响抗生素抗性基因的重要因素[41], 已有研究发现ARGs易在Proteobacteria、Firmicutes和Bacteroidetes之间转移, 尤其在Proteobacteria中, 其检测丰度较高[42].本研究通过网络分析发现, 与四环素类ARGs相关性较高的菌门主要是Proteobacteria、Firmicutes和Bacteroidota, 说明作为主要的与抗性基因相关的细菌门类, 这些门类的细菌可能是携带四环素类ARGs潜在宿主细菌的门类, 并且不同细菌门在共现性关系中起到了不同作用.其他学者也有相关发现, 如Awasthi等[43]研究发现Firmicutes和Proteobacteria是ARGs的宿主;有研究通过宏基因组学方法, 分析揭示了Firmicutes和Proteobacteria是较为常见的、潜在携带抗性基因的细菌宿主[44];以前的研究也揭示了在污水处理厂中一些常见的抗性微生物主要分属于Proteobacteria, 其次是Firmicutes和Bacteroidetes[45].除此之外, 本研究还发现一些节点数相对较少的门类, 如Acidobacteriota、Cyanobacteria、Spirochaetota和Myxococcota等, 尽管这些菌门在沉积物样品中丰度相对较低, 但是仍然发现与四环素类ARGs存在不同程度的共现性.
病原菌耐药性的传播, 使许多抗生素在治疗过程中失效, 继而引发人们过量用药的错误行为, 加重了病原菌耐药性的传播, 对人们的健康构成了严重威胁.病原菌通过基因水平转移获得ARGs是河流湿地生态系统安全最重要的风险因素之一, ARGs很容易被人类致病菌捕获, 从而形成超级细菌, 如沙门氏菌、拟杆菌、弯曲杆菌、志贺菌和大肠杆菌等[46].已有证据表明, 来自土壤的环境细菌与临床病原体之间存在ARGs交换, 例如两种高风险病原菌:肺炎克雷伯菌(Klebsiella pneumoniae)和鲍曼不动杆菌(Acinetobacter baumannii)[28, 47];Ju等[48]利用宏基因组分析, 在140个污水处理厂中观察到20种人类致病菌与ARGs共存.本研究中, 在检测到的27种病原菌中, 下游病原菌的丰度较上游增加了1.11%, 种类也更丰富.同时, 网络分析显示与病原菌显著相关的抗性基因主要来自下游新增的8种抗性基因, 占据了网络连接度的70%, 说明随着污染的输入, 下游沉积物中抗生素耐药基因在病原菌中的水平传播潜势要远远高于上游.同时, Liu等[13]研究也表明, 人类活动对携带ARGs的病原菌的相对丰度有显著影响, 城市化和大坝控制区携带ARGs的病原菌的相对丰度分别是原生环境的3.54倍和1.55倍.此外, 巴东氏菌(Bartonella henselae)、粪肠球菌(Enterococcus faecalis)、伯克氏菌(Burkholderia cenocepacia)、李斯特菌(Listeria monocytogenes)和普通拟杆菌(Bacteroides vulgatus)与四环素类AGRs相关性较高(同时与3个ARGs都相关), 表明这些病原菌可能是多个ARGs的潜在宿主;而百日咳博德特氏菌(Bordetella pertussis)、沙眼衣原体(Chlamydia trachomatis)和嗜肺军团菌(Legionella pneumophila)等与ARGs连接度较低的病原菌可能只携带单一ARGs.这些ARGs与病原菌之间可能存在转移和交换, 从而引发ARGs在病原菌之间的传播和扩散, 一旦抗性菌和ARGs与环境细菌、野生动物、家畜和人类接触, 通过HGT将ARGs转移给共生细菌和病原体, 将会引发高生态风险.
4 结论(1) 上、下游共检出15种四环素类ARGs, 其中上游检出7种, 下游检出13种, 上游沉积物中的四环素类ARGs含量约为下游的1.5倍.
(2) 四环素类抗生素与部分理化指标(如有效磷、总有机碳、硝态氮和总磷等)是影响四环素类ARGs分布的重要因素.
(3) 河流沉积物中共检出8 529条细菌OTUs序列, 分属64门、189纲、436目、686科、1 178属和2 615种.其中, 变形菌门(Proteobacteria)、厚壁菌门(Firmicutes)和拟杆菌门(Bacteroidota)是影响四环素类ARGs丰度的主要菌门.
(4) 共检出27种病原菌, 下游新增加的8种四环素类ARGs与病原菌有很高的相关性, 其中, 巴东氏菌(Bartonella henselae)、粪肠球菌(Enterococcus faecalis)、伯克氏菌(Burkholderia cenocepacia)、李斯特菌(Listeria monocytogenes)和普通拟杆菌(Bacteroides vulgatus)是影响四环素类ARGs传播的主要病原菌.
| [1] |
李奥林, 陈吕军, 张衍, 等. 抗生素抗性基因在两级废水处理系统中的分布和去除[J]. 环境科学, 2018, 39(10): 4593-4600. Li A L, Chen L J, Zhang Y, et al. Distribution and removal of antibiotic resistance genes in two sequential wastewater treatment plants[J]. Environmental Science, 2018, 39(10): 4593-4600. DOI:10.13227/j.hjkx.201801262 |
| [2] | Su Y L, Wang J X, Xia H P, et al. Comparative network analysis revealing the mechanisms of antibiotic resistance genes removal by leachate recirculation under different hydraulic loadings[J]. Science of the Total Environment, 2019, 649: 318-326. DOI:10.1016/j.scitotenv.2018.08.361 |
| [3] | Li L G, Huang Q, Yin X L, et al. Source tracking of antibiotic resistance genes in the environment-Challenges, progress, and prospects[J]. Water Research, 2020, 185. DOI:10.1016/j.watres.2020.116127 |
| [4] | Pei R T, Kim S C, Carlson K H, et al. Effect of river landscape on the sediment concentrations of antibiotics and corresponding antibiotic resistance genes (ARG)[J]. Water Research, 2006, 40(12): 2427-2435. DOI:10.1016/j.watres.2006.04.017 |
| [5] | Chee-Sanford J C, Aminov R I, Krapac I J, et al. Occurrence and diversity of tetracycline resistance genes in lagoons and groundwater underlying two swine production facilities[J]. Applied and Environmental Microbiology, 2001, 67(4): 1494-1502. DOI:10.1128/AEM.67.4.1494-1502.2001 |
| [6] | Allen H K, Donato J, Wang H H, et al. Call of the wild: antibiotic resistance genes in natural environments[J]. Nature Reviews Microbiology, 2010, 8(4): 251-259. DOI:10.1038/nrmicro2312 |
| [7] | D'Costa V M, King C E, Kalan L, et al. Antibiotic resistance is ancient[J]. Nature, 2011, 477(7365): 457-461. DOI:10.1038/nature10388 |
| [8] | Martínez J L. Antibiotics and antibiotic resistance genes in natural environments[J]. Science, 2008, 321(5887): 365-367. DOI:10.1126/science.1159483 |
| [9] |
林岚, 林琳, 陈恩中, 等. 宏基因组方法比较分析深海和珠江口沉积物中抗生素耐药基因的特征[J]. 中山大学学报(自然科学版), 2017, 56(2): 112-116. Lin L, Lin L, Chen E Z, et al. Metagemomic analysis on characteristics of antibiotic resistance genes in the deep ocean and Pearl River Estuary sediments[J]. Acta Scientiarum Naturalium Universitatis Sunyatseni, 2017, 56(2): 112-116. |
| [10] | Wang X P, Gong P, Wang C F, et al. A review of current knowledge and future prospects regarding persistent organic pollutants over the Tibetan Plateau[J]. Science of the Total Environment, 2016, 573: 139-154. DOI:10.1016/j.scitotenv.2016.08.107 |
| [11] |
李海萍, 王娜萍, 代宇庭. 云贵高原湿地景区人类活动强度的空间分布——以云南省拉市海流域为例[J]. 应用生态学报, 2021, 32(8): 2915-2922. Li H P, Wang N P, Dai Y T. Spatial distribution of human activity intensity in Yunnan-Guizhou Plateau Wetland scenicarea: a case study of Lashihai watershed in Yunnan Province, China[J]. Chinese Journal of Applied Ecology, 2021, 32(8): 2915-2922. DOI:10.13287/j.1001-9332.202108.017 |
| [12] |
臧泉. 武汉与西藏湖泊微生物群落多样性及抗生素抗性基因分布特征[D]. 拉萨: 西藏大学, 2021. Zang Q. Diversity of microbial community and distribution of antibiotic resistance genes in Lakes from Wuhan and Tibet[D]. Lhasa: Tibet University, 2021. |
| [13] | Liu S, Wang P F, Wang C, et al. Anthropogenic disturbances on antibiotic resistome along the Yarlung Tsangpo River on the Tibetan Plateau: ecological dissemination mechanisms of antibiotic resistance genes to bacterial pathogens[J]. Water Research, 2021, 202. DOI:10.1016/j.watres.2021.117447 |
| [14] |
罗晓, 张文丽, 袁立霞, 等. 纳污河流抗性基因和微生物群落相关性[J]. 中国环境科学, 2019, 39(6): 2606-2613. Luo X, Zhang W L, Yuan L X, et al. Correlation between resistance genes and microbial community in polluted rivers[J]. China Environmental Science, 2019, 39(6): 2606-2613. DOI:10.3969/j.issn.1000-6923.2019.06.044 |
| [15] | Ghernaout D, Elboughdiri N. Antibiotics resistance in water mediums: background, facts, and trends[J]. Applied Engineering, 2020, 4(1): 1-6. |
| [16] |
张文丽. 河流抗性基因传播与菌群特征时空演进规律分析[D]. 石家庄: 河北科技大学, 2019. Zhang W L. Analysis of temporal and spatial evolution laws of river resistance genes and flora characteristics propagation[D]. Shijiazhuang: Hebei University of Science and Technology, 2019. |
| [17] | Wang H, Li H Y, Gilbert J A, et al. Housefly larva vermicomposting efficiently attenuates antibiotic resistance genes in swine manure, with concomitant bacterial population changes[J]. Applied and Environmental Microbiology, 2015, 81(22): 7668-7679. DOI:10.1128/AEM.01367-15 |
| [18] | Wang H, Su X X, Su J Q, et al. Profiling the antibiotic resistome in soils between pristine and human-affected sites on the Tibetan Plateau[J]. Journal of Environmental Sciences, 2022, 111: 442-451. DOI:10.1016/j.jes.2021.04.019 |
| [19] |
姚鹏城, 陈嘉瑜, 张永明, 等. 废水处理系统中抗生素抗性基因分布特征[J]. 环境科学, 2019, 40(11): 5024-5031. Yao P C, Chen J Y, Zhang Y M, et al. Distribution characteristics of antibiotic resistance genes in wastewater treatment plants[J]. Environmental Science, 2019, 40(11): 5024-5031. DOI:10.13227/j.hjkx.201903196 |
| [20] |
夏飞扬, 马冬冬, 张军, 等. 会仙湿地典型河流抗生素污染特征及风险评价[J]. 桂林理工大学学报, 2021, 41(1): 174-182. Xia F Y, Ma D D, Zhang J, et al. Characteristics and risk assessment of typical antibiotic contamination in Huixian wetland rivers[J]. Journal of Guilin University of Technology, 2021, 41(1): 174-182. DOI:10.3969/j.issn.1674-9057.2021.01.022 |
| [21] |
于洁. 四环素对好氧活性污泥的抑制及对活性污泥四环素抗性的影响研究[D]. 天津: 南开大学, 2014. Yu J. Inhibition of tetracycline and impaction of tetracycline resistance in aerobic activated sludge[D]. Tianjin: Nankai University, 2014. |
| [22] |
宋现财. 四环素类抗生素在活性污泥上的吸附规律及其机理研究[D]. 天津: 南开大学, 2014. Song X C. Investigate the law of adsorption of tetracyclines on activated sludge and explore the mechanism[D]. Tianjin: Nankai University, 2014. |
| [23] |
苏建强, 黄福义, 朱永官. 环境抗生素抗性基因研究进展[J]. 生物多样性, 2013, 21(4): 481-487. Su J Q, Huang F Y, Zhu Y G. Antibiotic resistance genes in the environment[J]. Biodiversity Science, 2013, 21(4): 481-487. |
| [24] |
杨永青, 许继飞, 董泰音, 等. 水体和土壤环境中抗生素抗性基因(ARGs)的污染特征和消除[J]. 内蒙古农业科技, 2018, 46(3): 76-82. Yang Y Q, Xu J F, Dong T Y, et al. Pollution property and reduction of antibiotic resistance genes (ARGs) in aquatic and soil environment[J]. Journal of Northern Agriculture, 2018, 46(3): 76-82. DOI:10.3969/j.issn.2096-1197.2018.03.15 |
| [25] | Chen B W, Yang Y, Liang X M, et al. Metagenomic profiles of antibiotic resistance genes (ARGs) between human impacted estuary and deep ocean sediments[J]. Environmental Science & Technology, 2013, 47(22): 12753-12760. |
| [26] |
张丹丹, 郭亚平, 任红云, 等. 福建省敖江下游抗生素抗性基因分布特征[J]. 环境科学, 2018, 39(6): 2600-2606. Zhang D D, Guo Y P, Ren H Y, et al. Characteristics of antibiotic resistance genes in downstream areas of the Aojiang River, Fujian Province[J]. Environmental Science, 2018, 39(6): 2600-2606. |
| [27] | Allen H K, Moe L A, Rodbumrer J, et al. Functional metagenomics reveals diverse β-lactamases in a remote Alaskan soil[J]. The ISME Journal, 2009, 3(2): 243-251. DOI:10.1038/ismej.2008.86 |
| [28] | Forsberg K J, Reyes A, Wang B, et al. The shared antibiotic resistome of soil bacteria and human pathogens[J]. Science, 2012, 337(6098): 1107-1111. DOI:10.1126/science.1220761 |
| [29] | Storteboom H, Arabi M, Davis J G, et al. Tracking antibiotic resistance genes in the South Platte River basin using molecular signatures of urban, agricultural, and pristine sources[J]. Environmental Science & Technology, 2010, 44(19): 7397-7404. |
| [30] | Lin H, Chapman S J, Freitag T E, et al. Fate of tetracycline and sulfonamide resistance genes in a grassland soil amended with different organic fertilizers[J]. Ecotoxicology and Environmental Safety, 2019, 170: 39-46. |
| [31] |
吴楠, 乔敏. 土壤环境中四环素类抗生素残留及抗性基因污染的研究进展[J]. 生态毒理学报, 2010, 5(5): 618-627. Wu N, Qiao M. Tetracycline residues and tetracycline resistance gene pollution in soil: a review[J]. Asian Journal of Ecotoxicology, 2010, 5(5): 618-627. |
| [32] |
沈怡雯, 黄智婷, 谢冰. 抗生素及其抗性基因在环境中的污染、降解和去除研究进展[J]. 应用与环境生物学报, 2015, 21(2): 181-187. Shen Y W, Huang Z T, Xie B. Advances in research of pollution, degradation and removal of antibiotics and antibiotic resistance genes in the environment[J]. Chinese Journal of Applied and Environmental Biology, 2015, 21(2): 181-187. |
| [33] |
尤新新. 国内城市垃圾填埋场中抗生素残留与抗性基因的地域特征及理化指标关系研究[D]. 上海: 华东师范大学, 2018. You X X. The occurrence and distribution of antibiotics, antibiotic resistance genes and their relationships among physicochemical factors in landfill sites of Chinese Mainland[D]. Shanghai: East China Normal University, 2018. |
| [34] | Wu D, Huang Z T, Yang K, et al. Relationships between antibiotics and antibiotic resistance gene levels in municipal solid waste leachates in Shanghai, China[J]. Environmental Science & Technology, 2015, 49(7): 4122-4128. |
| [35] | Xu J, Xu Y, Wang H M, et al. Occurrence of antibiotics and antibiotic resistance genes in a sewage treatment plant and its effluent-receiving river[J]. Chemosphere, 2015, 119: 1379-1385. |
| [36] |
赵赛. 抗生素抗性基因在长江口滨岸沉积物中的赋存特征研究[D]. 上海: 华东师范大学, 2020. Zhao S. Occurrence and abundance of antibiotic resistance genes in sediments of the Yangtze River Estuary[D]. Shanghai: East China Normal University, 2020. |
| [37] | Guo X P, Pang W H, Dou C L, et al. Sulfamethoxazole and COD increase abundance of sulfonamide resistance genes and change bacterial community structures within sequencing batch reactors[J]. Chemosphere, 2017, 175: 21-27. |
| [38] | Di Cesare A, Eckert E M, Rogora M, et al. Rainfall increases the abundance of antibiotic resistance genes within a riverine microbial community[J]. Environmental Pollution, 2017, 226: 473-478. |
| [39] | McKinney C W, Loftin K A, Meyer M T, et al. tet and sul antibiotic resistance genes in livestock lagoons of various operation type, configuration, and antibiotic occurrence[J]. Environmental Science & Technology, 2010, 44(16): 6102-6109. |
| [40] | Wang Z, Han M Z, Li E H, et al. Distribution of antibiotic resistance genes in an agriculturally disturbed lake in China: their links with microbial communities, antibiotics, and water quality[J]. Journal of Hazardous Materials, 2020, 393. DOI:10.1016/j.jhazmat.2020.122426 |
| [41] | Zhou Z C, Zheng J, Wei Y Y, et al. Antibiotic resistance genes in an urban river as impacted by bacterial community and physicochemical parameters[J]. Environmental Science and Pollution Research, 2017, 24(30): 23753-23762. |
| [42] | Li D, Yu T, Zhang Y, et al. Antibiotic resistance characteristics of environmental bacteria from an oxytetracycline production wastewater treatment plant and the receiving river[J]. Applied and Environmental Microbiology, 2010, 76(11): 3444-3451. |
| [43] | Awasthi M K, Liu T, Chen H Y, et al. The behavior of antibiotic resistance genes and their associations with bacterial community during poultry manure composting[J]. Bioresource Technology, 2019, 280: 70-78. |
| [44] | Forsberg K J, Patel S, Gibson M K, et al. Bacterial phylogeny structures soil resistomes across habitats[J]. Nature, 2014, 509(7502): 612-616. |
| [45] | Liu Z B, Klümper U, Liu Y, et al. Metagenomic and metatranscriptomic analyses reveal activity and hosts of antibiotic resistance genes in activated sludge[J]. Environment International, 2019, 129: 208-220. |
| [46] | Fischbach M A, Walsh C T. Antibiotics for emerging pathogens[J]. Science, 2009, 325(5944): 1089-1093. |
| [47] | Perry J A, Wright G D. The antibiotic resistance "mobilome": searching for the link between environment and clinic[J]. Frontiers in Microbiology, 2013, 4. DOI:10.3389/fmicb.2013.00138 |
| [48] | Ju F, Li B, Ma L P, et al. Antibiotic resistance genes and human bacterial pathogens: Co-occurrence, removal, and enrichment in municipal sewage sludge digesters[J]. Water Research, 2016, 91: 1-10. |