 2021, Vol. 42
2021, Vol. 42



土壤微生物作为土壤生态系统的重要组成部分, 在土壤有机质分解、植物营养供给以及腐殖质形成过程中起着至关重要的作用.当环境发生变化时, 微生物的活动强度甚至群落组成都会产生相应地变化以适应新的环境[1~3], 因此, 可以用微生物群落的数量及组成结构来监测土壤环境的变化及生态系统恢复效果[4~7].
黄土高原作为典型退化生态系统的代表, 同时也是中国乃至世界重大的侵蚀区, 其生态环境治理和植被恢复是一项长期且艰巨的任务.自1999年起, 我国在黄土高原实施了退耕还林工程, 长期的植被恢复已经取得良好的宏观效果, 植被覆盖度与土壤质量都得到了不同程度的改善[8~10].目前已有较多学者针对黄土高原退耕还林区不同植被类型生态恢复过程, 围绕土壤水分特征、碳氮储量、土壤酶和土壤理化性质等开展了一系列研究[11~15], 结果表明不同植被类型对土壤性质有显著影响.而对于退耕地恢复过程中的植被与土壤、微生物群落的互动效应缺乏系统性的分析.本文特选择不同恢复阶段、不同模式人工林和天然次生林作为研究对象, 综合分析土壤微生物群落结构特征.
目前, 在黄土高原关于土壤微生物群落的研究主要采用的是传统的分离培养[16]、磷脂脂肪酸(PLFA)生物标记法[17]和高通量测序法[18].而高通量测序法能全面真实地描述微生物的群落特征, 目前, 该方法广泛用于揭示植被、作物栽培及土地管理方式、有机污染物、重金属、采煤沉陷、气候条件及其他方面等诸多因素对土壤微生物群落结构和生物量的影响[18~21].
基于此, 本文采用高通量测序技术, 以山西吉县森林生态系统国家野外科学观测研究站典型人工林(人工刺槐林和人工油松林)、天然次生林和草地为研究对象, 分析不同植被恢复模式下土壤细菌群落特征, 探讨细菌群落在植被恢复过程中对土壤生态系统的作用, 以期为黄土高原退耕还林区植被恢复的效益评价提供良好的数据基础, 并为人工林的合理经营提供科学依据.
1 材料与方法 1.1 研究区概况研究区位于山西吉县森林生态系统国家野外科学观测研究站(36.24~36.30°N, 110.66~110.79°E).属暖温带大陆性气候.多年平均年降水量579 mm, 年蒸发量1 729 mm, 年平均气温9.9℃, ≥10℃的积温3 358℃, 光照时数2 563.8 h, 无霜期172 d.地势东高西低, 海拔440~1 820 m, 黄河河谷最低.海拔1 350 m以下为典型黄土高原侵蚀地貌, 1 350 m以上为吕梁山脉土石山区.土壤主要为褐土, 森林植物地带属暖温带褐土阔叶林地带向森林草原地带的过渡地带.本区植物资源比较丰富, 天然植被主要有山杨(Popzrlus davidiana)、白皮松(Pinus bungeana)、榆树(Ulmus pumila L.)、华北落叶松(Larix principis-rupprechtii)、辽东栋(Quercus liaotungensis)、侧柏(Platycladus orientalis)、白桦(Betula platyphylla Suk.)、山桃[Prunus davidiana (Carr.) Franch.]、山杏(Pruns armeniaca varansu Maxim)、沙棘(Hippophae rhamnoides L.)和黄刺梅(Rosa xanthina Lindl.)等.人工植被主要有刺槐(Robinia psezrdoscacia L.)、油松(Pinus tabulaeformis Cam.)、侧柏(Platycladus orientalis)和沙棘(Hippophae rhamnoides)等.
1.2 土壤样品采集土壤样品2017年7月采集于山西吉县森林生态系统国家野外科学观测研究站.选取人工刺槐林、人工油松林、天然次生林和草地为研究对象, 样地详情见表 1.每种模式选择不同地理位置设置3个20 m×20 m的样地作为重复, 在设置的样地内, 按五点交叉取样法设置取样点; 去掉枯落物层后用土钻钻取表层0~20 cm土壤, 过2 mm筛后等量混合成一份土壤样品, 分装低温保存.回实验室后, 去掉土样中的石子、植物根和凋落物等杂物, 将土壤样品分为3份:一份迅速放入-80℃超低温冰箱中保存用于土壤微生物DNA提取; 一份土壤保存于4℃冰箱用于微生物量碳氮和pH值测定; 一份在阴凉处风干用于测定土壤化学计量等.
|
|
表 1 样地基本信息 Table 1 Description of sampling sites |
1.3 土壤样品测定 1.3.1 土壤微生物高通量测序
DNA提取:利用FastDNATM SPIN Kit for Soil试剂盒(MP Biomedicals, CA, USA)提取微生物基因组DNA, 用1%琼脂糖凝胶电泳检测抽提的基因组DNA的完整性, 超微量分光光度计(Nano Drop2000, USA)检测DNA浓度和纯度.PCR扩增:采用通用引物338F (5′-ACTCCTACGGGAGGCA GCAG-3′)和806R (5′-GGAGTACHVGGGTWTCTAA T-3′)对细菌的16S rRNA基因V4-V5区域进行PCR扩增[18].全部样本按照正式实验条件进行, 每个样本3个重复, 将同一样本的PCR产物混合后用2%琼脂糖凝胶电泳检测, 使用AxyPrepDNA凝胶回收试剂盒(Axygen Biosciences, Union City, CA, USA)切胶回收PCR产物, Tris_HCl洗脱; 2%琼脂糖电泳检测.测序:使用QuantiFluorTM-ST荧光计(Promega Biotech, Beijing, China)对PCR产物进行量化, 根据需要对样品进行测序调整, 然后利用Illumina MiSeq PE300平台(Shanghai Majorbio Bio-Pharm Technology Co., Ltd, China)进行高通量测序.
1.3.2 土壤基本特性分析土壤有机碳(TOC)、全氮(TN)、全磷(TP)、硝态氮(NO3--N)、铵态氮(NH4+-N)、速效磷(AP)和pH均按照文献[22]中的标准方法进行分析测定.
1.4 数据处理与分析 1.4.1 基本数据处理所有数据分析运用IBM SPSS Statistics Version 19、R version 4.0.3和美吉生物云平台(www.majorbio.com)进行处理.
1.4.2 OTU(operational taxonomic units)分析采用RDP classifier贝叶斯算法对97%相似水平的OTU代表序列进行分类学分析, 并分别在各个分类学水平:domain(域)、kingdom(界)、phylum(门)、class(纲)、order(目)、family(科)、genus(属)和species(种)统计各样本的群落物种组成.细菌16S rRNA对比数据库为Silva(Release132 http://www.arb-silva.de)和Greengene(Release 13.5 http://greengenes.secondgenome.com/).
1.4.3 稀释曲线(rarefaction curve)选择97%相似度的OTU, 利用mothur计算不同随机抽样下的α多样性指数, 利用R语言工具制作曲线图.
1.4.4 α多样性分析反映群落丰富度(community richness)的指数有Sobs、Chao和Ace.Chao和Ace指数是用来估算样本中所含OTU数目的指数, 是生态学中估计物种总数的常用指数之一, 但算法不同, 本次分析使用的计算公式分别见式(1)和式(2).
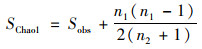
|
(1) |
式中, SChao1表示估计的OTU数, Sobs表示实际观测到的OTU数, n1表示只含有一条序列的OTU数目, n2表示只含有两条序列的OTU数目.
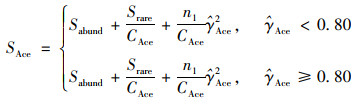
|
(2) |
式中:
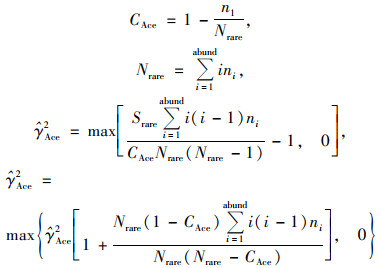
|
n1表示含有i条序列的OTU数目, Srare表示含有“abund”条序列或者少于“abund”的OTU数目, Sabund表示多于“abund”条序列的OTU数目, abund表示“优势”OTU的阈值, 默认为10.
反映群落多样性(community diversity)的指数有Shannon和Simpson.Shannon值越大, 说明群落多样性越高.Simpson指数值越大, 说明群落多样性越低.本次分析使用计算公式分别见式(3)和式(4).
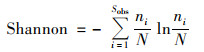
|
(3) |
式中, ni表示第i个OTU所含的序列数, N表示所有的序列数, Sobs表示实际观测到的OTUs.
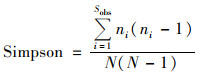
|
(4) |
式中各符号和字母含义同式(3).
反映群落覆盖度(community coverage)的指数是Coverage(C), 表示各样本的覆盖率, 其数值越高, 则样本中序列被测出的概率越高, 而没有被测出的概率越低.该指数反映本次测序结果是否代表了样本中微生物的真实情况.本次分析使用计算公式见式(5).
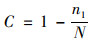
|
(5) |
式中, n1表示只含有一条序列的OTU数目, N表示抽样中出现的总序列数目.
1.4.5 β多样性分析本研究利用PCoA分析(principal co-ordinates analysis), 即主坐标分析, 基于Bray-Curtis距离算法, 对群落间的物种多样性进行组间比较分析, 探索不同分组样本间群落组成的相似性或差异性.
1.4.6 PICRUSt功能预测分析本研究采用PICRUSt软件进行菌群预测分析, 基于KEGG数据库(kyoto encyclopedia of genes and genomes).通过将基于Silva数据库的16S分类谱系转化为KEGG数据库中原核生物的分类谱系, 对16S RNA基因序列进行功能注释, 并根据OTU丰度计算各功能类别的丰度.
1.4.7 统计分析本研究采用冗余分析(redundancy analysis, RDA), 单因素方差分析(one-way ANOVA)并进行FDR多重检验校正, Pearson相关性系数分析等方法, Heatmap图采用Average层级类聚方式.
2 结果与分析 2.1 土壤基本特性不同恢复模式下土壤的基本特性如表 2所示. 5种土壤均呈弱碱性, pH值在8.46~8.69之间, 除草地外, 其他4种土壤之间差异不显著.TOC、TP和NH4+-N等3个指标在不同土壤中的含量差异显著, 其中CSL和CD的TOC含量要显著高于人工林(CH1、CH2和YS)中的含量, TP在CSL和CH1中的含量要显著低于在CH2、YS和CD中的含量, 而NH4+-N在不同恢复阶段的人工刺槐林(CH1和CH2)中的含量显著高于其他3种类型.TN、NO3--N和AP在5种土壤中的含量也存在差异性, 但差异不显著.
|
|
表 2 不同恢复模式下土壤的基本特性1) Table 2 Physicochemical properties of soil uner different vegetation restoration types |
2.2 高通量测序结果 2.2.1 OTU分析
通过V4-V5区测序, 按照最小样本序列数进行抽平, 并且保留至少在1个样本中的序列数都≥5的OTU, 5种样地的土壤样本共得到244 680条有效序列, 平均长度为438.99 bp, 采用RDP classifier贝叶斯算法对97%相似水平的OTU代表序列进行划分, 共得到25门、66纲、129类、240科、392属、760种和2 213 OTUs.
2.2.2 α多样性分析与稀释曲线表 3显示了测试样本的序列数及细菌群落的α多样性指数, 各样本的Coverage指数均大于98%, 说明土样中基因序列被检出的概率很高, 取样基本合理, 能够比较真实地反映土壤样本的细菌群落.从图 1(a)的Sobs指数稀释曲线可知, 随着随机抽取的测序数据量增加, Sobs指数曲线基本趋于平缓, 说明测序数据量合理, 更多的数据量只会产生少量新的物种.图 1(b)~1(f)显示多样性指数稀释曲线逐渐趋向平坦, 说明测序数据量足够大, 真实环境中细菌群落的置信度较高, 可以反映样本中绝大多数的微生物多样性信息.如表 3所示, 各样本Sobs、Ace和Chao 1指数差异性显著, 说明不同植被恢复模式下细菌群落的丰富度存在显著差异, 均表现为:CD>CSL>CH2>CH1>YS, 即草地和天然次生林细菌群落丰富度要大于人工林.人工林中, 刺槐林的细菌群落丰富度远大于油松林, 并且随着植被不断恢复, 土壤细菌群落的丰富度显著增加.而各模式之间细菌群落的多样性差异不显著, 说明土壤细菌种类没有明显的差异性.
|
|
表 3 测序统计及细菌群落多样性指数表 Table 3 Sequence statistics and diversity index of the bacterial communities |

|
选择97%相似度的OTU 图 1 多样性指数稀释曲线 Fig. 1 Rarefaction curves of diversity index |
在门和纲分类水平上统计各样本的物种丰度, 通过群落柱形和可视化圈可直观地研究群落组成, 反映每种植被恢复模式中优势物种分布比例, 以及各优势物种在不同植被恢复模式中的分布比例.如图 2所示, 在门分类水平上, 优势门类均为Actinobacteria(放线菌门)、Proteobacteria(变形菌门)、Acidobacteria (酸杆菌门)、Chloroflexi (绿弯菌门)、Gemmatimonadetes(芽单胞菌门)和Nitrospirae(硝化螺旋菌门), 约占到了所有微生物总数的90%以上, 但不同模式下各细菌群落优势种相对丰度有所差异, 但差异不显著.Actinobacteria的相对丰度变化为24.25%~28.79%(CD>CH1>CSL>YS>CH2), Proteobacteria为20.09%~28.63%(CSL>CH1>CH2>YS>CD), Acidobacteria为15.55%~19.87%(CH2>YS>CH1>CD>CSL), Chloroflexi为10.55%~17.88%(CD>CH2>YS>CH1>CSL), Gemmatimonadetes为3.51%~6.44%(YS>CH1>CSL>CH2>CD), Nitrospirae为3.04%~5.19%(CSL>CD>YS>CH1>CH2).在测试样本中, Bacteroidetes(拟杆菌门)和Firmicutes(厚壁菌门)的相对丰度均超过1%, 也是主要的土壤细菌门类.在门分类水平上, CSL和CD与人工林(CH1、CH2和YS)之间群落的相对丰度变化存在差异性, 但差异不显著.

|
(a)表示相对丰度, others表示所有样本中相对丰度低于1%的细菌物种; (b)表示可视化圈, 左半圈表示样本中物种组成情况, 外层彩带的颜色表示来自的分组, 内层彩带的颜色表示物种, 长度表示该物种在对应样本中的相对丰度; 右半圈表示在分类水平上物种在不同样本中的分布比例情况, 外层彩带表示物种, 内层彩带颜色表示不同分组, 长度表示该样本在某一物种中的分布比例, 下同 , 1.Actinobacteria; 2. Proteobacteria; 3.Acidobacteria; 4.Chloroflexi; 5.Gemmatimonadetes; 6.Nitrospirae; 7.Bacteroidetes; 8.others; 9.Firmicutes; 10.GAL15; 11.unclassified_k_norank; 12.Saccharibacteria; 13.Verrucomicrobia; 14.Tectomicrobia 图 2 在门分类水平上细菌群落相对丰度与可视化圈 Fig. 2 Relative abundance and circus diagram of bacterial community at the phylum level |
如图 3所示, 在纲分类水平上, 各恢复模式中的优势纲类均为Actinobacteria(放线菌纲)、Acidobacteria(酸杆菌纲)、Gemmatimonadetes(芽单胞菌纲)、Nitrospira(硝化螺旋菌纲)、α-Proteobacteria(α-变形菌纲)、β-Proteobacteria(β-变形菌纲)和δ-Proteobacteria(δ-变形菌纲), 约占到了所有微生物总数的74%以上, 而不同模式下各细菌群落优势种相对丰度也有所差异.Actinobacteria的相对丰度变化范围为24.25%~28.76%(CD>CH1>YS>CSL>CH2), Acidobacteria为15.55%~19.87%(CSL>CD>CH1>YS>CH2), Gemmatimonadetes为3.51%~6.44%(YS>CH1>CSL>CH2>CD), Nitrospira为3.04%~5.19%(CSL>CD>YS>CH1>CH2), α-Proteobacteria为10.35%~14.29%(CSL>CH1>CH2>YS>CD), β-Proteobacteria为4.10%~6.93%(CSL>CH2>CH1>YS>CD), δ-Proteobacteria为2.51%~4.38%(CSL>YS>CH1>CD>CH2).α-Proteobacteria、β-Proteobacteria和δ-Proteobacteria在CSL土壤中的相对丰度要高于其他4种恢复模式, 而在CD中的相对丰度最低.

|
1.Actinobacteria; 2.Acidobacteria; 3.α-Proteobacteria; 4.β-Proteobacteria; 5.Gemmatimonadetes; 6.Nitrospira; 7.others; 8.δ-Proteobacteria; 9.γ-Proteobacteria; 10. KD4-96; 11.Anaerolineae; 12.S085; 13.Chloroflexia; 14.Thermomicrobia; 15.Sphingobacteriia; 16.Bacilli; 17.TK10; 18.norank_p_GAL15; 19.Gitt-GS-136; 20.unclassified_k_norank; 21.norank_p_Saccharibacteria; 22.spartobacteria; 23.norank_p_Tectomicrobia 图 3 在纲分类水平上细菌群落相对丰度与可视化圈 Fig. 3 Relative abundance and circos diagram of bacterial community at the class level |
为了进一步对细菌群落物种进行组间和组内比较分析, 探索不同分组样本间群落组成的相似性或差异性, 特此进行了β多样性分析.基于Bray-Curtis距离算法, 采用PCoA分析衡量不同植被恢复模式下土壤细菌群落组间和组内物种多样性差异.如图 4所示, 在OTU水平上, PC1与PC2分别解释方差为24.46%和19.37%, 累计解释能力达43.83%, PC1可将CSL和YS的细菌群落与CH1、CH2和CD明显区分开, PC2可将YS和CH1与CSL、CH2和CD明显区分开, 影响群落结构的主导因子显著(P < 0.05).本研究发现, 各植被恢复模式组内样本间重复比较聚集, 土壤细菌群落物种多样性组间差异大于组内差异, CSL的土壤细菌群落物种多样性与人工林和CD明显分离.人工林中, CH1与CH2和YS之间存在分离, 但距离较小.

|
图 4 不同植被恢复模式土壤细菌群落主坐标分析(PCoA) Fig. 4 Principal coordinates analysis (PCoA) of soil bacteria with different vegetation restoration types, based on Bray-Curtis distances |
在门分类水平上, 对土壤中的优势细菌门类与土壤环境因子进行冗余分析(表 4和图 5), 结果表明, RDA1和RDA2分别解释对于物种数据影响程度的28.64%和8.36%, 累计解释量为37%.其中TN(RDA1=-0.942 6, RDA2=0.333 9, R2=0.603 6, P=0.005)和pH(RDA1=0.975 1, RDA2=-0.221 8, R2=0.535 9, P=0.013)对细菌群落结构的影响最大, 对细菌群落结构具有显著影响(P < 0.05).TN与Actinobacteria和Nitrospirae呈显著正相关, TOC与其也呈正相关关系, 但相关性不显著.pH与Proteobacteria和Gemmatimonadetes呈显著正相关, 与Actinobacteria呈显著负相关, 而Acidobacteria与TN和pH均呈显著负相关关系.
|
|
表 4 环境因子对RDA结果的解释权重 Table 4 Explanatory weights of environmental factors for RDA results |

|
TOC:土壤有机碳, TN:全氮, TP:全磷, NO3--N:硝态氮, NH4+-N:铵态氮, AP:速效磷, 下同; 蓝色箭头表示在门分类水平上优势细菌种群; 红色箭头表示环境因子 图 5 优势细菌门与土壤环境因子的冗余分析 Fig. 5 Redundancy analysis (RDA)of dominant bacterial phylum and soil environmental factors |
基于KEGG数据库预测的结果表明(表 5), 在一级功能层共获得6类生物代谢通路功能, 分别是细胞过程(cellular processes)、环境信息处理(environmental information processing)、遗传信息处理(genetic information processing)、人类疾病(human diseases)、代谢(metabolism)和有机系统(organismal systems).其中代谢、环境信息处理和遗传信息处理是一级功能层中最主要的组成部分, 占比分别为61.91%~62.49%、18.74%~19.28%和11.05%~11.31%.
|
|
表 5 一级功能层相对丰度/% Table 5 Relative abundance of KEGG pathway level 1/% |
同时对预测基因二级功能层丰度进行分析[图 6(a)], 发现各样本细菌群落主要涉及细胞运动(cell motility)、细胞内运输、分泌及囊泡运输(intracellular trafficking, secretion, and vesicular transport)、信号转导机制(signal transduction mechanisms)、氨基酸转运及代谢(amino acid transport and metabolism)和辅酶的运输及代谢(coenzyme transport and metabolism)等24个二级功能, 且不同植被恢复模式中细菌群落二级功能层OTU丰度间差异不显著, 均表现出功能上的丰富性.对所预测的二级功能层的OTU丰度和土壤环境因子进行Pearson相关性分析发现[图 6(b)], 绝大多数二级功能与土壤pH和TN存在显著的相关关系, 与土壤pH呈显著正相关关系, 与TN呈显著负相关关系.而对优势细菌门类和土壤环境因子的RDA分析结果也表明(图 5), 土壤pH和TN对细菌群落的影响最大, 对细菌群落结构具有显著影响.TP和NO3--N与部分二级功能也存在显著的相关关系.TOC和AP与二级功能存在负相关关系, NH4+-N与二级功能存在正相关关系, 但相关性均不显著.

|
(a)二级功能层丰度; (b)Pearson相关性热图, X轴和Y轴分别为环境因子和二级功能, 图例是表示不同R值的颜色区间, * 表示0.01 < P≤0.05, **表示0.001 < P≤0.01, ***表示P≤0.001; A:功能未知(function unknown), B:仅适用于一般功能预测(general function prediction only), C:氨基酸转运和代谢(amino acid transport and metabolism), D:能源生产和转化(energy production and conversion), E:信号转导机制(signal transduction mechanisms), F:转录(transcription), G:细胞壁/膜/包膜生物发生(cell wall/membrane/envelope biogenesis), H:碳水化合物的运输和代谢(carbohydrate transport and metabolism), I:无机离子的运输和代谢(inorganic ion transport and metabolism), J:复制、重组和修复(replication, recombination, and repair), K:翻译、核糖体结构与生物发生(translation, ribosomal structure, and biogenesis), L:脂质运输和代谢(lipid transport and metabolism), M:辅酶的运输和代谢(coenzyme transport and metabolism), N:翻译后修饰、蛋白质更新和保护蛋白(posttranslational modification, protein turnover, and chaperones), O:次生代谢物的生物合成、转运和分解代谢(secondary metabolites biosynthesis, transport, and catabolism), P:核苷酸的运输和代谢(nucleotide transport and metabolism), Q:防御机制(defense mechanisms), R:细胞内运输、分泌和囊泡运输(intracellular trafficking, secretion, and vesicular transport), S:细胞运动(cell motility), T:细胞周期控制、细胞分裂和染色体分区(cell cycle control, cell division, and chromosome partitioning), U:RNA加工和装饰(RNA processing and modification), V:染色质的结构和动力学(chromatin structure and dynamics), W:细胞骨架(cytoskeleton), Z:细胞外结构(extracellular structures) 图 6 二级功能层丰度和环境因子相关性热图 Fig. 6 One-way ANOVA and environmental factor correlation heatmap of KEGG pathway level 2 |
植被恢复主要通过影响枯落物的差异、根系形态和分泌物及整体生态系统物质和能量转化过程的差异来影响土壤微生物数量和群落, 微生物通过分解经过多年的累积枯枝落叶来影响土壤养分循环及其自身的结构和功能的多样性[23, 24].
有研究表明, Actinobacteria是降解木质素与纤维素的主要功能菌门, 本研究中5种植被恢复模式中Actinobacteria的相对丰度变化范围为24.25%~28.79%, 是群落中最优势细菌门, 这与刘洋等[25]对黄土高原不同乔木林土壤细菌群落特征的研究结果一致, 且植物群落类型是影响土壤放线菌多样性的重要因素[26].有研究表明, 在盐碱土中, Proteobacteria下的α-Proteobacteria、β-Proteobacteria、γ-Proteobacteria和δ-Proteobacteria是最重要的微生物纲类[27].在本研究中的不同植被恢复模式下, Proteobacteria相对丰度范围为20.09%~28.63%, 是土壤中主要的优势细菌门类, α-Proteobacteria是最优势纲, 其次是β-Proteobacteria, 相对丰度分别为10.35%~14.29%和4.10%~6.93%.且研究区土壤pH在8.46~8.69之间, 呈弱碱性, 也验证了Proteobacteria为碱性土壤中的主要优势群落.
植被恢复过程中, 植物枯落物增加了有机质向土壤的输入, 为微生物提供了充足的碳源和氮源, 对土壤微生物量产生了积极的影响.Zhang等[18]研究黄土高原不同时期(0、10、25和35 a)放牧后草原微生物群落的变化结果表明, 长期演替过程中, 植被和细菌的演替较土壤真菌的快, 植被通过改变土壤理化性质来改变土壤微生物群落的组成.Zeng等[28]选取了黄土高原从南到北的植被生态系统(森林、林草、草地、沙地和沙漠生态系统)作为研究对象, 研究了土壤性质对土壤微生物群落组成的影响, 发现Actinobacteria和Proteobacteria相对丰度与pH存在显著相关性, 说明pH对土壤细菌群落的影响较大.Xu等[29]研究了黄土高原土壤反硝化微生物群落对不同土地利用类型的响应, 结果表明, AN是影响微生物组成的关键环境因子.安丽芸等[30]研究了微生物多样性对土壤碳代谢特征的影响, 结果表明在黄土高原阔叶混交林, 土壤微生物多样性的降低显著影响了土壤的碳矿化速率和累计矿化量, 对其生态系统功能产生较大影响.而Zhao等[31]研究发现造林后土壤碳组分的变化对土壤微生物群落的多样性和结构有显著影响.以上研究均表明, 随着植被的不断恢复演替, 土壤养分和结构状况得到改善, 从而为土壤微生物的活动提供了良好的环境.微生物生物量总体增加和土壤酶活性不断增大使土壤肥力不断地得到恢复和提高, 改善了土壤微生物群落的结构和功能, 使得微生物群落从贫营养型向富营养型转变.
本研究表明草地和天然次生林土壤细菌群落丰富度要大于人工林.在人工林中, 刺槐林的土壤细菌群落丰富度远大于油松林, 并且随着植被不断恢复, 土壤细菌群落的丰富度明显增加.这与刘洋等[32]和Zhang等[33]的研究结果相似.Hu等[34]研究植被类型对黄土高原丘陵地区土壤微生物生物量和功能多样性的影响也表明, 对提高土壤微生物量碳氮含量而言, 林草混合模式比单一植被的效果更好, 能有效促进土壤修复.以上研究均表明混交模式造林对于土壤质量改善作用效果最好, 刺槐纯林次之, 荒草地和油松纯林较差.故建议当地退耕还林应以混交林为主, 对提高土壤肥力和质量效果最好.曾全超等[35]的研究证明了在黄土丘陵区, 人工刺槐林对土壤碳氮库的增加有一定的作用, 但是相对于辽东栎和侧柏等天然次生林有一定的差距.以刺槐等为主的植物群落, 其根部共生的根瘤菌可以将空气中的无机氮转化为有机氮固定于土壤中, 进而改变根区土壤系统的物质组成和肥力水平[36].Liu等[37]的研究表明, 与原生草地相比, 人工刺槐林显著提高了土壤有机碳、全氮、碳氮比和碳磷比, Vítková等[38]也报道了刺槐种植后由于共生根际细菌的固氮能力导致了较高的氮浓度.封晔等[39]对黄土高原8种植物根际细菌与丛枝菌根真菌群落多样性及其相互关系进行了研究, 结果表明植物种类和根际环境对根际微生物群落结构有较大影响, 同时, 刺槐的根际细菌和AMF群落多样性指数均较高, 因此刺槐可作为黄土高原区植被恢复的先锋树种.
4 结论(1) 不同植被恢复模式下土壤细菌群落的丰富度存在显著差异, 均表现为:CD>CSL>CH2>CH1>YS, 即草地和天然次生林土壤细菌群落丰富度要大于人工林.人工林中, 刺槐林的土壤细菌群落丰富度远大于油松林, 并且随着植被不断恢复, 土壤细菌群落的丰富度明显增加.
(2) 各植被恢复模式下土壤细菌优势门类均为: Actinobacteria、Proteobacteria、Acidobacteria、Chloroflexi、Gemmatimonadetes和Nitrospirae, 优势纲类均为: Actinobacteria、Acidobacteria、α-Proteobacteria、β-Proteobacteria和δ-Proteobacteria.CSL和CD与人工林(CH1、CH2和YS)之间土壤细菌群落的相对丰度变化存在差异性.
(3) 各植被恢复模式下, 土壤优势细菌门类与TN和pH具有显著相关性, 说明在土壤环境因子中TN和pH对土壤细菌群落的影响较大.其中TN与Actinobacteria和Nitrospirae呈显著正相关, pH与Proteobacteria、Gemmatimonadetes呈显著正相关, 与Actinobacteria呈显著负相关, 而Acidobacteria与TN和pH均呈显著负相关关系.
(4) 代谢、环境信息处理和遗传信息处理是黄土高原退耕还林区土壤细菌群落一级功能层中最主要的组成部分.主要涉及氨基酸转运和代谢、能源生产和转化、信号转导机制、转录、碳水化合物的运输和代谢、无机离子的运输和代谢等24个二级功能, 均表现出功能上的丰富性, 且绝大多数二级功能与土壤pH、TN存在显著的相关关系.
致谢: 感谢朱金兆老师、房世鹏同学、张英俊同学和山西吉县生态站工作人员对野外工作的支持. 感谢上海美吉生物医药科技有限公司和中国科学院植物研究所植被与环境变化国家重点实验室提供的样品检测支持.
| [1] | Wardle D A, Walker L R, Bardgett R D. Ecosystem properties and forest decline in contrasting long-term chronosequences[J]. Science, 2004, 305(5683): 509-513. DOI:10.1126/science.1098778 |
| [2] | Luo C Y, Zhang B X, Liu J, et al. Effects of different Ages of Robinia pseudoacacia plantations on soil physiochemical properties and microbial communities[J]. Sustainability, 2020, 12(21). DOI:10.3390/su12219161 |
| [3] |
李娜, 王宝荣, 安韶山, 等. 黄土高原草地土壤细菌群落结构对于降水变化的响应[J]. 环境科学, 2020, 41(9): 4284-4293. Li N, Wang B R, An S S, et al. Response of soil bacterial community structure to precipitation change in grassland of Loess Plateau[J]. Environmental Science, 2020, 41(9): 4284-4293. |
| [4] | Zhalnina K, Dias R, de Quadros P D, et al. Soil pH determines microbial diversity and composition in the park grass experiment[J]. Microbial Ecology, 2015, 69(2): 395-406. DOI:10.1007/s00248-014-0530-2 |
| [5] | Nacke H, Goldmann K, Schöning I, et al. Fine spatial scale variation of soil microbial communities under European beech and Norway spruce[J]. Frontiers in Microbiology, 2016, 7. DOI:10.3389/fmicb.2016.02067 |
| [6] | Pei Z Q, Eichenberg D, Bruelheide H, et al. Soil and tree species traits both shape soil microbial communities during early growth of Chinese subtropical forests[J]. Soil Biology and Biochemistry, 2016, 96: 180-190. DOI:10.1016/j.soilbio.2016.02.004 |
| [7] | Ding X L, Zhang B, Lü X X, et al. Parent material and conifer biome influence microbial residue accumulation in forest soils[J]. Soil Biology and Biochemistry, 2017, 107: 1-9. DOI:10.1016/j.soilbio.2016.12.020 |
| [8] |
尤南山, 董金玮, 肖桐, 等. 退耕还林还草工程对黄土高原植被总初级生产力的影响[J]. 地理科学, 2020, 40(2): 315-323. You N S, Dong J W, Xiao T, et al. The effects of the "grain for green" project on gross primary productivity in the Loess Plateau[J]. Scientia Geographica Sinica, 2020, 40(2): 315-323. |
| [9] |
山仑, 徐炳成. 新时期黄土高原退耕还林(草)有关问题探讨[J]. 水土保持通报, 2019, 39(6): 295-297. Shan L, Xu B C. Discussion on some issues about returning farmland to forest or grassland on Loess Plateau in new era[J]. Bulletin of Soil and Water Conservation, 2019, 39(6): 295-297. |
| [10] |
谭学进, 穆兴民, 高鹏, 等. 黄土区植被恢复对土壤物理性质的影响[J]. 中国环境科学, 2019, 39(2): 713-722. Tan X J, Mu X M, Gao P, et al. Effects of vegetation restoration on changes to soil physical properties on the loess plateau[J]. China Environmental Science, 2019, 39(2): 713-722. DOI:10.3969/j.issn.1000-6923.2019.02.034 |
| [11] | Kou M, Jiao J Y, Yin Q L, et al. Successional trajectory over 10 years of vegetation restoration of abandoned slope croplands in the Hill-Gully region of the Loess Plateau[J]. Land Degradation & Development, 2016, 27(4): 919-932. |
| [12] | Feng X M, Fu B J, Piao S L, et al. Revegetation in China's Loess Plateau is approaching sustainable water resource limits[J]. Nature Climate Change, 2016, 6(11): 1019-1022. DOI:10.1038/nclimate3092 |
| [13] |
周俊杰, 陈志飞, 杨全, 等. 黄土丘陵区退耕草地土壤呼吸及其组分对氮磷添加的响应[J]. 环境科学, 2020, 41(1): 479-488. Zhou J J, Chen Z F, Yang Q, et al. Response of soil respiration and its components to nitrogen and phosphorus addition in farming-withdrawn grassland in the semiarid loess hilly-gully region[J]. Environmental Science, 2020, 41(1): 479-488. |
| [14] |
刘俊廷, 张建军, 孙若修, 等. 晋西黄土区退耕年限对土壤孔隙度等物理性质的影响[J]. 北京林业大学学报, 2020, 42(1): 94-103. Liu J T, Zhang J J, Sun R X, et al. Effects of the conversion time of cropland into forestry on soil physical properties in loess area of western Shanxi Province of northern China[J]. Journal of Beijing Forestry University, 2020, 42(1): 94-103. |
| [15] |
瞿晴, 徐红伟, 吴旋, 等. 黄土高原不同植被带人工刺槐林土壤团聚体稳定性及其化学计量特征[J]. 环境科学, 2019, 40(6): 2904-2911. Qu Q, Xu H W, Wu X, et al. Soil aggregate stability and its stoichiometric characteristics in Robinia pseudoacacia forest within different vegetation zones on the Loess Plateau, China[J]. Environmental Science, 2019, 40(6): 2904-2911. |
| [16] |
邢肖毅, 黄懿梅, 安韶山, 等. 黄土丘陵区不同植被土壤氮素转化微生物生理群特征及差异[J]. 生态学报, 2013, 33(18): 5608-5614. Xing X Y, Huang Y M, An S S, et al. Characteristics of physiological groups of soil nitrogen-transforming microbes in different vegetation types in the Loess Gully region, China[J]. Acta Ecologica Sinica, 2013, 33(18): 5608-5614. |
| [17] |
魏安琪, 魏天兴, 刘海燕, 等. 黄土区刺槐和油松人工林土壤微生物PLFA分析[J]. 北京林业大学学报, 2019, 41(4): 88-98. Wei A Q, Wei T X, Liu H Y, et al. PLFA analysis of soil microorganism under Robinia pseudoacacia and Pinus tabuliformis plantation in loess area[J]. Journal of Beijing Forestry University, 2019, 41(4): 88-98. |
| [18] | Zhang C, Liu G B, Song Z L, et al. Interactions of soil bacteria and fungi with plants during long-term grazing exclusion in semiarid grasslands[J]. Soil Biology and Biochemistry, 2018, 124: 47-58. DOI:10.1016/j.soilbio.2018.05.026 |
| [19] |
李娜英, 韩智勇, 王双超, 等. 多污染源作用下填埋场地下水微生物群落分析[J]. 中国环境科学, 2020, 40(11): 4900-4910. Li N Y, Han Z Y, Wang S C, et al. Impacts of different pollution sources on the microbial community in groundwater at municipal solid waste landfill sites[J]. China Environmental Science, 2020, 40(11): 4900-4910. DOI:10.3969/j.issn.1000-6923.2020.11.031 |
| [20] |
马静, 卢永强, 张琦, 等. 黄土高原采煤沉陷对土壤微生物群落的影响[J]. 土壤学报, 2020. Ma J, Lu Y Q, Zhang Q, et al. Effects of coal mining subsidence on soil microbial community in the Loess Plateau[J]. Acta Pedologica Sinica, 2020. DOI:10.11766/trxb202003160122 |
| [21] |
王娜, 高婕, 魏静, 等. 三江平原湿地开垦对土壤微生物群落结构的影响[J]. 环境科学, 2019, 40(5): 2375-2381. Wang N, Gao J, Wei J, et al. Effects of wetland reclamation on soil microbial community structure in the Sanjiang plain[J]. Environmental Science, 2019, 40(5): 2375-2381. |
| [22] | 鲍士旦. 土壤农化分析[J]. 北京: 中国农业出版社, 2000. |
| [23] | Okubo A, Matsusaka M, Sugiyama S. Impacts of root symbiotic associations on interspecific variation in sugar exudation rates and rhizosphere microbial communities: a comparison among four plant families[J]. Plant and Soil, 2016, 399(1-2): 345-356. DOI:10.1007/s11104-015-2703-2 |
| [24] | Spohn M, Widdig M. Turnover of carbon and phosphorus in the microbial biomass depending on phosphorus availability[J]. Soil Biology and Biochemistry, 2017, 113: 53-59. DOI:10.1016/j.soilbio.2017.05.017 |
| [25] |
刘洋, 曾全超, 黄懿梅. 基于454高通量测序的黄土高原不同乔木林土壤细菌群落特征[J]. 中国环境科学, 2016, 36(11): 3487-3494. Liu Y, Zeng Q C, Huang Y M. Soil microbial communities by 454prosequencing under different arbor forests on the Loess Plateau[J]. China Environmental Science, 2016, 36(11): 3487-3494. DOI:10.3969/j.issn.1000-6923.2016.11.035 |
| [26] |
张晓红, 胡文革, 莫超, 等. 艾比湖湿地根际放线菌多样性及其环境响应[J]. 环境科学与技术, 2015, 38(12): 22-30, 134. Zhang X H, Hu W G, Mo C, et al. Correlation of Actinobacteria community diversity in three different rhizospheres and physicochemical properties in natural Reserve of Ebinur Lake wetland[J]. Environmental Science & Technology, 2015, 38(12): 22-30, 134. |
| [27] |
林耀奔, 杨建辉, 叶艳妹. 盐碱地不同土地利用方式下土壤细菌群落结构多样性差异分析[J]. 环境科学学报, 2019, 39(4): 1266-1273. Lin Y B, Yang J H, Ye Y M. Analysis on diversity of soil bacterial community under different land use patterns in saline-alkali land[J]. Acta Scientiae Circumstantiae, 2019, 39(4): 1266-1273. |
| [28] | Zeng Q C, Dong Y H, An S S. Bacterial community responses to soils along a latitudinal and vegetation gradient on the Loess Plateau, China[J]. PLoS One, 2016, 11(4). DOI:10.1371/journal.pone.0152894 |
| [29] | Xu Y D, Zhong Z K, Zhang W, et al. Responses of soil nosZ-type denitrifying microbial communities to the various land-use types of the Loess Plateau, China[J]. Soil and Tillage Research, 2019, 195. DOI:10.1016/j.still.2019.104378 |
| [30] |
安丽芸, 李君剑, 严俊霞, 等. 微生物多样性对土壤碳代谢特征的影响[J]. 环境科学, 2017, 38(10): 4420-4426. An L Y, Li J J, Yan J X, et al. Effects of microbial diversity on soil carbon mineralization[J]. Environmental Science, 2017, 38(10): 4420-4426. |
| [31] | Zhao F Z, Ren C J, Zhang L, et al. Changes in soil microbial community are linked to soil carbon fractions after afforestation[J]. European Journal of Soil Science, 2018, 69(2): 370-379. DOI:10.1111/ejss.12525 |
| [32] |
刘洋, 黄懿梅, 曾全超. 黄土高原不同植被类型下土壤细菌群落特征研究[J]. 环境科学, 2016, 37(10): 3931-3938. Liu Y, Huang Y M, Zeng Q C. Soil bacterial communities under different vegetation types in the Loess Plateau[J]. Environmental Science, 2016, 37(10): 3931-3938. |
| [33] | Zhang C, Liu G B, Xue S, et al. Soil bacterial community dynamics reflect changes in plant community and soil properties during the secondary succession of abandoned farmland in the Loess Plateau[J]. Soil Biology and Biochemistry, 2016, 97: 40-49. DOI:10.1016/j.soilbio.2016.02.013 |
| [34] | Hu C J, Fu B J, Liu G H, et al. Vegetation patterns influence on soil microbial biomass and functional diversity in a hilly area of the Loess Plateau, China[J]. Journal of soils and sediments, 2010, 10(6): 1082-1091. DOI:10.1007/s11368-010-0209-3 |
| [35] |
曾全超, 李鑫, 董扬红, 等. 黄土高原不同乔木林土壤微生物量碳氮和溶解性碳氮的特征[J]. 生态学报, 2015, 35(11): 3598-3605. Zeng Q C, Li X, Dong H Y, et al. Soil microbial biomass nitrogen and carbon, water soluble nitrogen and carbon under different arbors forests on the Loess Plateau[J]. Acta Ecologica Sinica, 2015, 35(11): 3598-3605. |
| [36] |
翟辉, 张海, 邱梅, 等. 黄土高原退耕坡地不同类型林分土壤生物学活性的研究[J]. 西北林学院学报, 2016, 31(4): 33-38, 72. Zhai H, Zhang H, Qiu M, et al. Soil biological activity of different tree types on slope land converted from farmland in the Loess Plateau[J]. Journal of Northwest Forestry University, 2016, 31(4): 33-38, 72. DOI:10.3969/j.issn.1001-7461.2016.04.06 |
| [37] | Liu J L, Ngoc Ha V, Shen Z, et al. Response of the rhizosphere microbial community to fine root and soil parameters following Robinia pseudoacacia L. afforestation[J]. Applied Soil Ecology, 2018, 132: 11-19. DOI:10.1016/j.apsoil.2018.08.004 |
| [38] | Vítková M, Tonika J, Müllerová J. Black locust-Successful invader of a wide range of soil conditions[J]. Science of the Total Environment, 2015, 505: 315-328. DOI:10.1016/j.scitotenv.2014.09.104 |
| [39] |
封晔, 唐明, 陈辉, 等. 黄土高原六道沟流域8种植物根际细菌与AMF群落多样性研究[J]. 环境科学, 2012, 33(1): 314-322. Feng Y, Tang M, Chen H, et al. Community diversity of bacteria and arbuscular mycorrhizal fungi in the rhizosphere of eight plants in Liudaogou watershed on the Loess Plateau China[J]. Environmental Science, 2012, 33(1): 314-322. |