 2021, Vol. 42
2021, Vol. 42



2. 南京信息工程大学环境科学与工程学院, 江苏省大气环境监测与污染控制高技术研究重点实验室, 南京 210044
2. School of Environmental Science and Engineering, Nanjing University of Information Science & Technology, Jiangsu Key Laboratory of Atmospheric Environment Monitoring and Pollution Control, Nanjing 210044, China
气溶胶粒子经过一系列光物理和化学过程, 对气候、空气质量和人类健康都产生重要影响[1~3].据估计约有104~105种VOCs在经历大气光化学反应后产生一系列的氧化产物[4~6], 有助于二次有机气溶胶(secondary organic aerosol, SOA)的形成.液相(云滴、雾滴和气溶胶水)中的光化学反应是SOA的另一个重要生成途径[7], 有些反应过程(如酸催化和齐聚化反应)只发生在凝聚相[8].液相反应通过特定的反应机制改变气溶胶的化学组成, 尤其是近年来提出液相SOA(aqSOA)是大气中吸光性有机物——棕色碳(BrC)的潜在来源[9~11].有研究表明[12~15], 气溶胶粒子经历液相氧化反应后, 生成的新粒子在波长300~400 nm处吸光性增强.
已知的aqSOA相关参数主要通过实验室研究和模式模拟手段得到, 与大气气溶胶中前体物反应环境还有一定差距.例如, Kaur等[16]的研究表明, 加利福尼亚冬季PM2.5水溶性组分光照下会形成·OH、1O2和3C*等自由基, 进而影响实际气溶胶水的液相反应历程; 有研究表明咪唑类物质液相中可以氧化异戊二烯形成aqSOA[17], 而真实环境中由于产生的3C*自由基浓度低并没有观察到aqSOA的形成[18], 说明实验室单一模拟物质液相氧化结果不能完全反映实际大气真实环境条件.目前, 大部分研究只关注模拟VOCs/SVOCs/IVOCs(挥发性/半挥发性/中等挥发性有机物)液相氧化aqSOA特性和反应机制[8, 19, 20], 实际大气液相体系中上述前体物降解产物特性的研究还很少.例如, Bateman等[21]进行了溶解在云滴中的SOA(通过d-柠檬烯臭氧暗反应形成)的液相氧化; Zhao等[20]研究了生物质燃料燃烧产生的BrC液相光化学氧化后产物的吸光性变化.实际大气有机组分液相氧化研究偏少的一个重要原因是产物复杂, 产物定性定量较困难.
高分辨飞行时间气溶胶质谱仪(HR-ToF-AMS)等先进的技术手段结合气相色谱质谱(GC-MS)和高效液相色谱质谱(HPLC-MS)等分子水平的测定, 可以实现对混合产物氧化性和元素组成等的表征.Lee等[22]采用HR-ToF-AMS对比了实际气溶胶水和云水液相反应SOA的形成; 本课题组前期也已采用上述方式对四乙基愈创木酚等单一前体物液相光化学氧化反应开展了研究[23~25].为说明大气实际液相体系中溶解性有机质(dissolved organic matter, DOM)的光化学降解, 本文连续采集了20 d实际大气PM2.5滤膜, 通过测定有机碳、元素碳和主要水溶性离子等, 选取PM2.5及对应的上述组分浓度相近的8张膜进行实验.PM2.5滤膜溶解在超纯水中过滤萃取制备成反应溶液(DOM), 考察了不同光源直接光解和·OH氧化光解4种体系中DOM的光化学反应过程, 采用黑炭-气溶胶质谱仪(SP-AMS)和UV-vis表征降解产物随光照时间的氧化特性和吸光性的变化, 评估实际大气中液相反应对环境和气候的影响.
1 材料与方法 1.1 DOM反应液制备采样点位于常州市区西南部, 江苏理工学院57号楼9层楼楼顶(离地面大约40 m), 紧邻京杭大运河和中吴大道, 周围为居民区, 无建筑物遮挡.采用大流量采样器以1.05 m3·min-1流量采集PM2.5样品到石英滤膜(20.3 cm×25.4 cm, Whatman QM-A)上.采样时间2018年5月1~20日, 每天采集23 h.采样前, 将石英滤膜置于500℃马弗炉中焙烧4 h, 采样前后, 放置在恒温恒湿干燥箱(温度22℃, 湿度45%)中平衡48 h, 用精度为0.01 mg的天平称重, 然后用铝箔封装储存在-20℃低温冰柜中保存.
将1/2 PM2.5滤膜用超纯水(电阻率: 18.2 MΩ·cm)超声波振荡提取45 min, 取出摇匀, 用一次性针头过滤器(孔径0.45 μm, 直径13 mm)过滤, 加超纯水定容至600 mL, 配置成初始TOC浓度约为10 mg·L-1的DOM, 置于光化学反应仪中进行光化学反应.·OH氧化实验时, 另外加入H2O2以产生·OH.
1.2 仪器和试剂大流量采样器(KB-1000型, 青岛金仕达电子科技有限公司); UV光化学反应仪(XPA-7型, 南京胥江机电厂); 模拟太阳光光化学反应仪(RPR-200型, 美国Rayonet公司); 紫外可见分光光度计(UV-vis, Specord 210 plus, 德国耶拿); 总有机碳分析仪(TOC, TOC-LCPII, 日本岛津); 离子色谱分析仪(IC, Aquion, 美国赛默飞); 高效液相色谱(HPLC, LC-2030C 3D, 日本岛津); 蒸发光散射仪(ELSD-LT Ⅱ, 日本岛津); 黑炭-气溶胶质谱仪(SP-AMS, 美国Aerodyne公司); 紫外辐射照度计(U-20型, 杭州远方光电信息股份有限公司).
硫酸铵、过氧化氢、氢氧化钾、邻-苯二甲酸氢钾、碳酸氢钠和碳酸钠购自Alfa Aesar公司; 甲醇(色谱级)购自Fisher公司; 有机酸标准品购自Sigma-Aldrich公司; 盐酸(优纯级)购自国药集团.
1.3 实验装置和过程紫外光降解: 功率为500 W的汞灯, 发射光谱波长为284、297、313、340、365和404 nm, 具体实验装置和辐射光谱见文献[22], 反应液表面UVB(290~320nm)辐射为6257.1 μW·cm-2.光解流程: 汞灯放在冷阱中, 冷阱的中空部分通入冷凝水对灯源进行冷却.上述制备的600 mL DOM平均倒入6根石英反应管中, 其中1根反应管用锡箔纸包裹作为暗反应对照组.反应仪底部有磁力搅拌装置, 转子以1 200 r·min-1的转速搅拌以保证溶液混合均匀.
模拟太阳光降解: 首次采用文献[26, 27]建议的模拟太阳光, 组合300、350和419 nm灯源(分别为2、7和7根), 装置如图 1所示, 具体辐射特性见文献[26, 27].光解流程: 将DOM混合均匀倒入30根石英反应管(20 mL), 并插入光化学反应仪内的样品托盘上.反应管内放入搅拌转子, 利用压缩空气源带动磁力搅拌装置, 使反应溶液连续搅拌均匀.反应器底部装有冷却风扇对灯管和反应液进行降温, 使反应控制在25~30℃的条件下进行.同样设置1根用锡箔纸包裹的暗反应对照样品.

|
图 1 光化学反应仪(模拟太阳光)实验装置 Fig. 1 Experimental apparatus for photo-chemical reaction (simulated solar light) |
本文进行了4个反应体系的实验研究: 太阳光、太阳光+·OH、紫外光和紫外光+·OH.在不同反应阶段(初始光照1 h取样, 之后每隔2 h取样, 累计23 h)取出一定体积的溶液分别进行后续的离线分析.由于每张膜溶解的溶液约1 200 mL只能进行两次实验, 为了保证每个体系进行3次平行实验, 取2张膜溶解制备约2 400 mL的DOM溶液, 每次取600 mL进行光解实验, 取3次实验的平均值, 标准偏差标注在后续实验结果上.
1.4 分析方法 1.4.1 吸光性测定取约2 mL反应液于1 cm石英比色皿中, 利用UV-vis对波长范围200~600 nm进行光谱扫描, 测定产物在不同波长处的吸光度.
1.4.2 DOM降解率分析取约15 mL反应液于TOC进样瓶中, 测得总碳和无机碳, 差值即为TOC浓度(本文用水溶性有机碳即WSOC表示), 用WSOC的降解率表征DOM的降解率.
1.4.3 SP-AMS分析反应溶液使用雾化器进行雾化处理, 进入硅胶干燥器, 随后通过SP-AMS进行分析.SP-AMS工作的基本原理[28]: 使用特制的空气动力学透镜, 将环境空气中的细粒子聚集成束, 粒子再经热分解(600℃)和电子轰击离子化(70 eV), 最后进入飞行时间质谱仪得到其质谱(MS谱).根据SP-AMS所测得的有机物MS谱图数据可以获得DOM的元素组成(氧碳比, O/C; 氢碳比, H/C)及有机质与有机碳比(OM/OC)等.根据文献[29], 利用aqSOA的AMS半定量方法, 以Org/SO42-比值量化aqSOA的生成量变化.
1.4.4 棕色碳(HULIS浓度)分析HLB固相萃取柱预先加入3 mL甲醇和3 mL酸化超纯水(用HCl将pH调至2)进行活化平衡.取出15 mL反应液酸化(pH=2)后倒入固相萃取柱进行过滤, 加入2 mL超纯水淋洗, 随后向柱内加入3 mL 2%氨水/甲醇(体积比)洗脱出HULIS, 洗脱液氮吹干后定容至1.5 mL的液相小瓶中, 利用HPLC-ELSD对HULIS进行定量分析.HPLC的操作条件: 流动相为乙腈∶水=20∶80(体积比), 流速为0.6 mL·min-1, 进样体积为60 μL. ELSD的操作条件: N2以1.5 L·min-1的流速和漂移管温度以(80~90℃)运行, GAIN设定为6. HULIS标准曲线通过5个浓度梯度(0、1、5、10、25和50 mg·L-1)的萨旺泥河黄腐酸(SRFA, 国际腐殖质协会)标准溶液获得.
1.4.5 有机酸分析取5 mL反应液于IC进样瓶, 采用自动进样的方式用IC分析.IC分析条件: Dionex公司的IonPac AS11-HC型分离柱(250 mm×4 mm)和IonPac AG11型保护柱(50 mm×4 mm), 15 mmol·L-1 KOH溶液作为淋洗液, 流速1.2 mL·min-1.
2 结果与讨论 2.1 吸光性分析图 2为4个体系DOM随光照时间变化的UV-vis谱图及MAE随光照时间的变化. 其中,
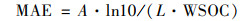
|

|
(a1)和(a2)太阳光; (b1)和(b2) 太阳光+·OH; (c1)和(c2)紫外光; (d1)和(d2) 紫外光+·OH; 箭头表示趋势, 双向箭头表示先下降后升高 图 2 降解产物随光照时间变化的平均UV-vis谱图和不同波长处的单位质量吸收效率 Fig. 2 Average UV-vis spectra of degradation products and unit mass absorption efficiency at different wavelengths for different reaction time |
式中,A为波长为λ处的吸光度, L为光程长度(0.01 m), WSOC为测定的溶液TOC值, mg·L-1.
从图 2可知, 在波长200~600 nm范围内均没有特征吸收峰出现, DOM的吸光度随着波长的增加而减小. 值得说明的是, 由于4个体系原始DOM对应的PM2.5滤膜不完全一致, 初始吸光度稍微有所差异. 外加H2O2后由于未反应的H2O2在200~250 nm有一定的吸收, 导致初始溶液吸光度也有所升高. 图 2(a1)和2(b1)太阳光反应体系中, 随光照时间的延长, 吸光度变化不明显;相反, 图 2(c1)和2(d1)UV体系中吸光度随光照时间延长不断下降, 这可能与UV具有更高的辐射强度有关. 相同光源下, ·OH氧化反应体系中, ·OH加速了前体物的降解, 碎片化反应生成CO2或者小分子VOCs挥发到大气中, 使得吸光度下降趋势较明显[图 2(b1)和2(d1)]. 图 2(a2)~2(d2)为MAE与波长的关系.
随着光照时间的延长, 两个紫外光体系中MAE值都单调升高[图 2(c2)和2(d2)], MAE值随着光照时间延长升高说明大气液相反应是产生生色团的重要途径, 与Lee等[22]的研究结论一致.而太阳光体系MAE值先下降后升高, 与紫外光照时变化趋势不一致, 说明降解机制有一定差异.
2.2 DOM降解率分析图 3为4个体系溶液WSOC浓度与光照时间的关系.从中可知, WSOC随光照时间延长不断降低, 且UV光照下更明显, 说明DOM中有机质不断被降解.

|
图 3 WSOC浓度和aqSOA相对质量(Org/SO42-)随反应时间变化 Fig. 3 Changes in WSOC concentrations and aqSOA relative mass (Org/SO42-) with reaction time |
采用SP-AMS进一步分析了DOM的降解(以Org/SO42-表示).如图 3(b)所示, 由于4个体系实验用的膜不完全一致(SO42-离子浓度在5~12 μg·m-3), 导致初始溶液的Org/SO42-会有点差异.然而有研究表明[30], 云水中无机盐(SO42-等离子)对降解几乎没有影响, 因此初始Org/SO42-不同不影响后面讨论的光解变化趋势.由图 3(b)可知, DOM最终光解的低挥发性有机物含量均有所下降, 太阳光、太阳光+·OH、紫外光和紫外光+·OH这4个体系中, 光照23 h时Org/SO42-分别下降了18.1%、58.3%、76.5%和80.8%, 紫外光体系Org/SO42-下降明显高于太阳光体系, 与WSOC变化规律一致.Lee等[22]采用实际膜萃取有机物的液相氧化过程研究也发现Org/SO42-随光照时间延长而降低; Org/SO42-下降进一步说明DOM液相氧化过程中发生了碎片化反应, 有机物含量不断降低, 这与很多模拟单一物质液相氧化过程中aqSOA含量不断升高的结果正好相反[23, 24].这可能是因为实际膜的DOM中含有供电子化合物(例如吡啶、咪唑等)和HULIS光敏性物质等, 发生氧化还原反应促进了体系ROS的形成[31].Kroll等[32]的研究也表明, 羟基等含氧官能团的加入, 使得挥发性产物不断增多, 进而扩散到大气环境中, 导致Org/SO42-下降.
2.3 产物氧化特性碳氧化态(OSc=2O/C-H/C)被广泛用于描述aqSOA氧化性和老化程度[22].图 4为DOM光降解后aqSOA的O/C、H/C和OSc与光照时间的关系.从中可知, 太阳光和太阳光+·OH这2个体系, O/C、H/C和OSc随光照时间有所变化, 但变化不明显, 其变化范围分别在0.3~0.5、1.5~1.7和-1.0~-0.2之间.与单一前体物降解时H/C、O/C随反应时间的延长分别下降和升高明显不同; 紫外光和紫外光+·OH这2个体系中, O/C和OSc随反应时间的延长有所下降, H/C没有明显的变化规律, 说明UV条件下DOM降解了, 这个与图 3中UV条件下WSOC不断下降的结论一致.单一前体物UV光解的研究中[8], O/C和OSc也随光照时间而下降, 但下降幅度明显高于本文实际的DOM, 说明实际DOM中多个共组分存在使得光解机制更加复杂.值得说明的是, AMS检测到有机产物只是低挥发性产物, 而一些含氧产物, 例如甲酸(O/C=2)极易挥发, 雾化时已经挥发, 未被AMS检测到, 因此AMS测定的产物的O/C不能代表所有降解产物的O/C.

|
图 4 不同光照时间下aqSOA的元素比(O/C、H/C)和OSc Fig. 4 Elemental ratios(O/C, H/C)and OSc of aqSOA at different illumination times |
根据Alien等[33]建立的相关性关系, f44值可作为估算环境有机气溶胶(OA)O/C的另一个指标.因此, 本文进一步绘制了aqSOA在不同反应时间下的f44和f43三角图(f44和f43分别代表质核比(m/z)44和43的有机物占总OA的质量分数), 如图 5所示, 其中虚线所示的空间三角形边界区域为大气OA中f44和f43分布的区域[34].根据SOA的氧化程度, OA又可以分为低挥发氧化性OA(LV-OOA)和半挥发氧化性OA(SV-OOA).太阳光体系中产物f44值比较低, 主要聚集在SV-OOA区域, 而太阳光+·OH体系f44值先增加后减小, 产物氧化程度与LV-OOA相似, 与文献[29]中云水有机物氧化反应初始阶段产物先增加30%, 随后在氧化后期又出现下降的结果相似.UV和UV+·OH反应体系, 由于光子能量高, f44值呈现下降的趋势, 其中UV+·OH由于·OH的强氧化作用, f44随光照时间单调下降, 说明反应过程中DOM不断降解成小分子有机酸和CO2等产物.

|
图 5 不同光照时间下aqSOA的f44和f43三角图 Fig. 5 The f44 vs. f43 triangle plot of aqSOA at different illumination times |
棕色碳是一类吸光性含C物质的总称, HULIS作为BrC的一个重要组成部分, 由于C=C和C=O双键的基团中的π-π*电子跃迁, 在紫外区(300~400 nm)吸光性强, 可见光区吸光性弱[35].而大气混合气溶胶在酸催化条件下发生的均相和非均相反应被认为是形成HULIS最为重要的途径[36]. Lambe等[12]的研究表明, 大气中aqSOA的形成过程对HULIS有显著的贡献作用, 其贡献大小取决于前体物类型及其氧化水平.为此, 本研究分析了反应过程中HULIS的浓度变化, 进一步说明反应过程中吸光性的变化. 图 6为7个反应时间点(0、1、7、11、15、19和23 h)生成的HULIS浓度变化.从中可知: 反应初始阶段溶液中HULIS浓度为4.49 mg·L-1.太阳光照反应条件下, HULIS浓度波动很小, 说明基本不形成新的HULIS物质, 这与太阳光照体系中吸光度变化不大相吻合(图 2).而UV和UV+·OH条件下, 随反应时间的延长, HULIS浓度成倍数增长, 并且生成速率在反应后期不断加快, 反应23h时浓度高达17.84 mg·L-1, 说明DOM中高分子量物质不断分解或氧化为含羧基、羟基和芳香基等官能团的物质.

|
图 6 光照过程生成HULIS浓度与反应时间的关系 Fig. 6 Relationship between HULIS concentration and reaction time during illumination |
草酸、丁二酸和丙二酸等二元酸在对流层大气颗粒物中普遍存在[37].有研究表明[38], 一元或二元羧酸是大气中甲基乙二醛[36]和苯酚类化合物[24]的液相氧化产物, 不同羧酸之间不断进行转化.例如, 甲基乙二醛大气液相氧化形成乙二酸和丙酮酸[37]; 丙二酸大气液相·OH氧化生成羟基丙二酸[37].根据反应机制[37~39], 液相·OH氧化时, 各种酸之间发生以下转化: 乙醇酸(乙酸, 丙酮酸)→乙醛酸→草酸; 乙酸→甲醛→甲酸; 丙二酸→丁二酸→苹果酸.为此, 本文定量了7种有机酸的浓度, 结果见图 7.太阳光体系中没有检测到苹果酸, 所以只有6种.太阳光和太阳光+·OH体系中, 乙醛酸和草酸的生成量具有相同的变化趋势.草酸作为连锁反应的最终产物, 4个体系中草酸的生成量最高, 且最高值基本都出现在15~19 h之间.甲酸作为云雾中重要的产物[40], 在本文中浓度并不是很高, 与本课题组以前酚类化合物·OH氧化光解时主要生成甲酸的结论不一致[24], 说明实际DOM组分大气液相氧化与单一物质对有机酸的生成有显著不同.UV+·OH体系下, 由于产生的·OH更多, 实际膜中部分前体物降解为羰基、羧基和羟基等官能团化合物, 这些物质进一步转化为有机酸, 导致有机酸浓度稍微高于UV体系.

|
图 7 有机酸生成量随光照时间变化 Fig. 7 Changes in organic acids production with illumination time |
然而, 由于实际膜成分的复杂性, 有机酸的生成量与光解时间之间没有明显的关联性.同时, 由于本文采用IC定量有机酸, 精度偏低, 低浓度的有机酸很难检测到, 今后需要借助精度更高的仪器例如离子色谱-电喷雾质谱(IC-ESI-MS)进行更详细的测定.但是本文的研究首次直接证实实际大气混合气溶胶液相氧化会生成多种羧酸, 并在粒子中积累, 成为大气aqSOA中必不可少的一部分.
3 结论(1) 紫外光体系中DOM得到不断降解, 相应的产物f44值远低于太阳光体系; 太阳光、太阳光+·OH两个体系, O/C、H/C和OSc随光照时间有所变化, 但变化不明显, 其变化范围分别在0.3~0.5、1.5~1.7和-1.0~-0.2之间.
(2) 太阳光反应体系中, 随光照时间的延长, 降解后溶液的吸光度变化不明显, 而UV体系中吸光度随光照时间延长不断下降; 紫外光体系中MAE值随光照时间单调升高, 说明大气液相反应是产生生色团的重要途径, 而太阳光体系MAE值先下降后升高, 与UV光照时变化趋势不一致, 说明降解机制有一定差异.
(3) 太阳光照反应条件下基本不形成新的HULIS物质, 随光照时间延长溶液吸光度变化也不大; 而UV和UV+·OH条件下, 随反应时间的延长, HULIS浓度成倍数增长, 反应23 h时HULIS浓度为初始的4倍, 说明DOM中部分高分子量物质不断分解或氧化为含羧基、羟基和芳香基等官能团的物质.4个体系中, 都检测到多种羧酸, 其中草酸的生成量最高, 且最高值基本都出现在15~19 h之间.
| [1] | Hallquist M, Wenger J C, Baltensperger U, et al. The formation, properties and impact of secondary organic aerosol: current and emerging issues[J]. Atmospheric Chemistry and Physics, 2009, 9(14): 5155-5236. DOI:10.5194/acp-9-5155-2009 |
| [2] | Ervens B, Turpin B J, Weber R J. Secondary organic aerosol formation in cloud droplets and aqueous particles (aqSOA): a review of laboratory, field and model studies[J]. Atmospheric Chemistry and Physics, 2011, 11(21): 11069-11102. DOI:10.5194/acp-11-11069-2011 |
| [3] | Ye Z L, Li Q, Ma S S, et al. Summertime day-night differences of PM2.5 components (Inorganic ions, OC, EC, WSOC, WSON, HULIS, and PAHs) in Changzhou, China[J]. Atmosphere, 2017, 8(10). DOI:10.3390/atmos8100189 |
| [4] | George C, Ammann M, D'Anna B, et al. Heterogeneous photochemistry in the atmosphere[J]. Chemical Reviews, 2015, 115(10): 4218-4258. DOI:10.1021/cr500648z |
| [5] | Lu J C, Ge X L, Liu Y, et al. Significant secondary organic aerosol production from aqueous-phase processing of two intermediate volatility organic compounds[J]. Atmospheric Environment, 2019, 211: 63-68. DOI:10.1016/j.atmosenv.2019.05.014 |
| [6] |
叶招莲, 瞿珍秀, 马帅帅, 等. 气溶胶水相反应生成二次有机气溶胶研究进展[J]. 环境科学, 2018, 39(8): 3954-3964. Ye Z L, Qu Z X, Ma S S, et al. Secondary organic aerosols from aqueous reaction of aerosol water[J]. Environmental Science, 2018, 39(8): 3954-3964. |
| [7] |
陈彦彤, 李旭东, 陶冶, 等. 有机激发三重态参与的光化学反应研究进展[J]. 化工进展, 2020, 39(8): 3344-3353. Chen Y T, Li X D, Tao Y, et al. A review on photochemical reaction via organic triplet excited state[J]. Chemical Industry and Engineering Progress, 2020, 39(8): 3344-3353. |
| [8] | Li Y J, Huang D D, Cheung H Y, et al. Aqueous-phase photochemical oxidation and direct photolysis of vanillin-a model compound of methoxy phenols from biomass burning[J]. Atmospheric Chemistry and Physics, 2014, 14(6): 2871-2885. DOI:10.5194/acp-14-2871-2014 |
| [9] | Xu J, Cui T Q, Fowler B, et al. Aerosol brown carbon from dark reactions of syringol in aqueous aerosol mimics[J]. ACS Earth and Space Chemistry, 2018, 2(6): 608-617. DOI:10.1021/acsearthspacechem.8b00010 |
| [10] | Nozière B, Dziedzic P, Córdova A. Formation of secondary light-absorbing "fulvic-like" oligomers: a common process in aqueous and ionic atmospheric particles?[J]. Geophysical Research Letters, 2007, 34(21). DOI:10.1029/2007GL031300 |
| [11] | Chang J L, Thompson J E. Characterization of colored products formed during irradiation of aqueous solutions containing H2O2 and phenolic compounds[J]. Atmospheric Environment, 2010, 44(4): 541-551. DOI:10.1016/j.atmosenv.2009.10.042 |
| [12] | Lambe A T, Cappa C D, Massoli P, et al. Relationship between oxidation level and optical properties of secondary organic aerosol[J]. Environmental Science & Technology, 2013, 47(12): 6349-6357. |
| [13] | Hoffer A, Kiss G, Blazsó M, et al. Chemical characterization of humic-like substances (HULIS) formed from a lignin-type precursor in model cloud water[J]. Geophysical Research Letters, 2004, 31(6). DOI:10.1029/2003GL018962 |
| [14] |
闫才青, 郑玫, 张远航. 大气棕色碳的研究进展与方向[J]. 环境科学, 2014, 35(11): 4404-4414. Yan C Q, Zheng M, Zhang Y H. Research progress and direction of atmospheric brown carbon[J]. Environmental Science, 2014, 35(11): 4404-4414. |
| [15] |
蔡竟, 支国瑞, 陈颖军, 等. 中国秸杆焚烧及民用燃煤棕色碳排放的初步研究[J]. 环境科学研究, 2014, 27(5): 455-461. Cai J, Zhi G R, Chen Y J, et al. A preliminary study on brown carbon emissions from open agricultural biomass burning and residential coal combustion in China[J]. Research of Environmental Sciences, 2014, 27(5): 455-461. |
| [16] | Kaur R, Labins J R, Helbock S S, et al. Photooxidants from brown carbon and other chromophores in illuminated particle extracts[J]. Atmospheric Chemistry and Physics, 2019, 19(9): 6579-6594. DOI:10.5194/acp-19-6579-2019 |
| [17] | Aregahegn K Z, Nozière B, George C. Organic aerosol formation photo-enhanced by the formation of secondary photosensitizers in aerosols[J]. Faraday Discussions, 2013, 165: 123-134. DOI:10.1039/c3fd00044c |
| [18] | Tsui W G, Rao Y, Dai H L, et al. Modeling photosensitized secondary organic aerosol formation in laboratory and ambient aerosols[J]. Environmental Science & Technology, 2017, 51(13): 7496-7501. |
| [19] | Renard P, Siekmann F, Salque G, et al. Aqueous-phase oligomerization of methyl vinyl ketone through photooxidation-Part 1: aging processes of oligomers[J]. Atmospheric Chemistry and Physics, 2015, 15(1): 21-35. DOI:10.5194/acp-15-21-2015 |
| [20] | Zhao R, Lee A K Y, Huang L, et al. Photochemical processing of aqueous atmospheric brown carbon[J]. Atmospheric Chemistry and Physics, 2015, 15(11): 6087-6100. DOI:10.5194/acp-15-6087-2015 |
| [21] | Bateman A P, Nizkorodov S A, Laskin J, et al. Photolytic processing of secondary organic aerosols dissolved in cloud droplets[J]. Physical Chemistry Chemical Physics, 2011, 13(26): 12199-12212. DOI:10.1039/c1cp20526a |
| [22] | Lee A K Y, Hayden K L, Herckes P, et al. Characterization of aerosol and cloud water at a mountain site during WACS 2010: secondary organic aerosol formation through oxidative cloud processing[J]. Atmospheric Chemistry and Physics, 2012, 12(15): 7103-7116. DOI:10.5194/acp-12-7103-2012 |
| [23] |
庄雨, 陈彦彤, 李旭东, 等. 四乙基愈创木酚液相·OH氧化SOA产率及特征分析: 初始浓度的影响[J]. 环境科学, 2020, 41(1): 146-154. Zhuang Y, Chen Y T, Li X D, et al. Secondary organic aerosol mass yield and characteristics from 4-ethylguaiacol aqueous·OH oxidation: effects of initial concentration[J]. Environmental Science, 2020, 41(1): 146-154. |
| [24] | Ye Z L, Zhuang Y, Chen Y T, et al. Aqueous-phase oxidation of three phenolic compounds by hydroxyl radical: Insight into secondary organic aerosol formation yields, mechanisms, products and optical properties[J]. Atmospheric Environment, 2020, 223. DOI:10.1016/j.atmosenv.2019.117240 |
| [25] | Ye Z L, Qu Z X, Ma S S, et al. A comprehensive investigation of aqueous-phase photochemical oxidation of 4-ethylphenol[J]. Science of the Total Environment, 2019, 685: 976-985. DOI:10.1016/j.scitotenv.2019.06.276 |
| [26] | Smith J D, Sio V, Yu L, et al. Secondary organic aerosol production from aqueous reactions of atmospheric phenols with an organic triplet excited state[J]. Environmental Science & Technology, 2014, 48(2): 1049-1057. |
| [27] | George K M, Ruthenburg T C, Smith J, et al. FT-IR quantification of the carbonyl functional group in aqueous-phase secondary organic aerosol from phenols[J]. Atmospheric Environment, 2015, 100: 230-237. DOI:10.1016/j.atmosenv.2014.11.011 |
| [28] |
江琪, 王飞, 孙业乐. 河北香河亚微米气溶胶组分特性、来源及其演变规律分析[J]. 环境科学, 2018, 39(7): 3022-3032. Jiang Q, Wang F, Sun Y L. Analysis of chemical composition, source and evolution of submicron particles in Xianghe, Hebei Province[J]. Environmental Science, 2018, 39(7): 3022-3032. |
| [29] | Lee A K Y, Zhao R, Gao S S, et al. Aqueous-phase OH oxidation of glyoxal: application of a novel analytical approach employing aerosol mass spectrometry and complementary off-line techniques[J]. The Journal of Physical Chemistry A, 2011, 115(38): 10517-10526. DOI:10.1021/jp204099g |
| [30] | Boris A J, Desyaterik Y, Collett J L. How do components of real cloud water affect aqueous pyruvate oxidation?[J]. Atmospheric Research, 2014, 143: 95-106. DOI:10.1016/j.atmosres.2014.02.004 |
| [31] | Dou J, Lin P, Kuang B Y, et al. Reactive oxygen species production mediated by humic-like substances in atmospheric aerosols: enhancement effects by pyridine, imidazole, and their derivatives[J]. Environmental Science & Technology, 2015, 49(11): 6457-6465. |
| [32] | Kroll J H, Seinfeld J H. Chemistry of secondary organic aerosol: formation and evolution of low-volatility organics in the atmosphere[J]. Atmospheric Environment, 2008, 42(16): 3593-3624. DOI:10.1016/j.atmosenv.2008.01.003 |
| [33] | Aiken A C, DeCarlo P F, Kroll J H, et al. O/C and OM/OC ratios of primary, secondary, and ambient organic aerosols with high-resolution time-of-flight aerosol mass spectrometry[J]. Environmental Science & Technology, 2008, 42(12): 4478-4485. |
| [34] | Ng N L, Canagaratna M R, Zhang Q, et al. Organic aerosol components observed in northern hemispheric datasets from aerosol mass spectrometry[J]. Atmospheric Chemistry and Physics, 2010, 10(10): 4625-4641. DOI:10.5194/acp-10-4625-2010 |
| [35] |
顾远, 李清, 黄雯倩, 等. 常州市冬季PM2.5中类腐殖质昼夜特征分析[J]. 环境科学, 2019, 40(3): 1091-1100. Gu Y, Li Q, Huang W Q, et al. Day-night characteristics of humic-like substances in PM2.5 during winter in Changzhou[J]. Environmental Science, 2019, 40(3): 1091-1100. |
| [36] |
项萍, 谭吉华, 马永亮, 等. 大气颗粒物中类腐殖酸的研究进展[J]. 环境化学, 2015, 34(3): 401-409. Xiang P, Tan J H, Ma Y L, et al. Research progress of humic-like substances (HULIS) in atmospheric particles[J]. Environmental Chemistry, 2015, 34(3): 401-409. |
| [37] | Herrmann H, Tilgner A, Barzaghi P, et al. Towards a more detailed description of tropospheric aqueous phase organic chemistry: CAPRAM 3.0[J]. Atmospheric Environment, 2005, 39(23-24): 4351-4363. DOI:10.1016/j.atmosenv.2005.02.016 |
| [38] | Tan Y, Carlton A G, Seitzinger S P, et al. SOA from methylglyoxal in clouds and wet aerosols: measurement and prediction of key products[J]. Atmospheric Environment, 2010, 44(39): 5218-5226. DOI:10.1016/j.atmosenv.2010.08.045 |
| [39] | Altieri K E, Seitzinger S P, Carlton A G, et al. Oligomers formed through in-cloud methylglyoxal reactions: chemical composition, properties, and mechanisms investigated by ultra-high resolution FT-ICR mass spectrometry[J]. Atmospheric Environment, 2008, 42(7): 1476-1490. DOI:10.1016/j.atmosenv.2007.11.015 |
| [40] | Chameides W L, Davis D D. Aqueous-phase source of formic acid in clouds[J]. Nature, 1983, 304(5925): 427-429. DOI:10.1038/304427a0 |