 2021, Vol. 42
2021, Vol. 42



2. 中国地质大学(武汉)环境学院大气科学系, 武汉 430074
2. Department of Atmospheric Sciences, School of Environmental Studies, China University of Geosciences, Wuhan 430074, China
大气颗粒物可通过吸收和散射太阳辐射影响地球热平衡, 还能直接影响大气能见度、区域灰霾污染的形成和人体健康[1~3]. 有机气溶胶是大气颗粒物的重要组分, 在我国诸多地区其平均质量浓度占总颗粒物的(15±5)%[4]. 黑碳(black carbon, BC)是气溶胶中最主要的吸光组分, 主要来源于生物质和化石燃料燃烧[5]. 有机碳(organic carbon, OC)的吸光作用在大气模式研究中常被忽略[6], 但大量研究表明具有吸光作用的OC(棕碳, brown carbon, BrC)在源排放和大气颗粒物中普遍存在[7~10]. 典型BrC的吸光范围包括近紫外和可见光区域(300~550 nm), 其吸光作用对波长有很强的依赖性[11]. 因BrC吸光作用导致的增温效应约占大气中总碳质气溶胶的20%~24%[12~14].
根据现有研究, 生物质燃烧是BrC的主要一次来源[5, 11]. 煤和石油等化石燃料燃烧以及大气中的多相反应过程也是BrC的重要来源[15~19]. 我国有关BrC吸光性质的研究主要集中在京津冀[20, 21]、珠江三角洲[22]和关中平原[23]等区域的城市地带. 这些研究均发现生物质燃烧对BrC的吸光作用有显著影响. 尽管南京及周边地区的家庭能源主要为电力和天然气, 且秸秆焚烧已于2014年被全面禁止, 生物质燃烧排放仍可通过大气传输影响该地区BrC的吸光作用. 已有研究表征了南京北郊BrC的吸光作用[24], 但目前对该地区BrC的来源和时间变化规律仍缺乏深入认识. 为进一步了解我国东部地区BrC的吸光特征和来源, 本研究分析了南京北郊2018-09~2019-09 PM2.5的化学组成及有机组分的吸光作用, 并结合主成分分析判定BrC的主要来源, 以期为探究和细化我国东部地区大气BrC的来源提供参考.
1 材料与方法 1.1 样品采集采样点位于南京信息工程大学图书馆楼顶(32.203024 °N, 118.713513 °E), 距地面约35 m, 周边主要为居住区和交通干道, 东北方向约5 km处为化工园区. 采用PM2.5-PUF-300型大气采样器(广州铭野)以300 L·min-1的流速将PM2.5收集于18 cm×11 cm的石英纤维滤膜上(MK360, 瑞典Munktell). 从2018-09-28~2019-09-28每6 d采集一次, 每次采样分为19:00~07:00(次日, 12 h)和08:00~19:00(11 h)两个时间段. 滤膜在采样前于550℃下烘烤5 h, 冷却后称重(QUINTIX125D-1CN天平, 德国Sartorius)待用. 采样期间共收集110个PM2.5滤膜样品和8个场地空白样品, 在控温控湿环境中(20~25℃, RH≈50%)平衡24 h后称重并密封保存在-20℃条件下.
1.2 化学组成和吸光性质分析PM2.5样品中的OC和EC采用热/光碳分析仪(2001A, 美国DRI)在IMPROVE-A方法条件下定量. 分别用超纯水(18.25 MΩ·cm)和甲醇(HPLC级)提取PM2.5中具有吸光作用的有机组分, 并测量其吸收光谱, 具体分析过程如下.
水提取:取1/4滤膜(49.5 cm2)在40 mL超纯水中超声萃取30 min, 然后用孔径为0.22 μm的聚四氟乙烯亲水性滤头(上海安谱)过滤. 采用紫外可见分光光度计(UV-1900, 日本岛津)在200~900 nm的波长(λ)范围内测量水溶性有机碳(water-soluble organic carbon, WSOC)的吸收光谱. 样品溶液中的WSOC采用总有机碳分析仪(TOC-LCPH/CPN, 日本岛津)测定, 水溶性无机离子组分(NH4+、NO3-、SO42-、K+、Mg2+和Ca2+)采用离子色谱仪(ICS-2000, ICS-3000, 美国Dionex)分析.
甲醇提取:取6 cm2滤膜在10 mL甲醇中超声萃取30 min并过滤(疏水性滤头). 甲醇可提取有机碳(methanol extractable organic carbon, MEOC)的吸收光谱测定范围和WSOC相同. 将经甲醇提取后的滤膜在通风橱中晾干, 采用热/光碳分析仪以同样的方法测定残留的OC. 以上所有分析过程均扣除场地空白.
1.3 吸光性质特征参数计算样品水提取液的吸光度(Aλ, w)通过公式(1)转换为光吸收系数(Absλ, w, Mm-1)[25]:
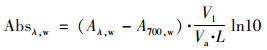
|
(1) |
式中, A700, w代表基线漂移, Vl为萃取溶剂体积(m3), Va是样品滤膜对应的采样体积(m3), L是光程长度(0.01 m). 质量吸收效率(mass absorption efficiency, MAEλ, w, m2·g-1)可用来表征吸光组分的吸收效率[10], 其计算公式(2)为:
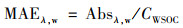
|
(2) |
式中, CWSOC表示样品中WSOC的质量浓度(μg·m-3). 埃氏吸收指数(absorption Ångström exponent, Å)常用于表征BrC吸光作用对波长的依赖程度. 本研究中ÅWSOC由lg Absλ, w与lg λ在波长300~550 nm的线性回归斜率确定. 水对OC的提取效率(ηw, %)用公式(3)计算:
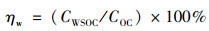
|
(3) |
式中, COC为OC的质量浓度(μg·m-3).
对样品的甲醇提取溶液, 光吸收系数(Absλ, m, Mm-1)和ÅMEOC的计算方法和水提取液相同. MEOC的质量浓度CMEOC(μg·m-3)用公式(4)计算:
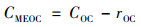
|
(4) |
式中, rOC是甲醇提取后滤膜上残留OC的质量浓度(μg·m-3). 质量吸收效率(MAEλ, m, m2·g-1)由公式(5)计算:

|
(5) |
甲醇对OC的提取效率(ηm, %)由公式(6)计算:
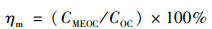
|
(6) |
本研究采用365 nm处的吸光特征参数代表BrC的吸光性质. 已有研究表明BrC在该波长处具有显著的吸光作用, 而无机组分(硝酸盐等)的干扰可忽略[25]. 另外, 以往研究重点关注BrC在365 nm的吸光特征[18, 20, 23, 26], 选择该波长有利于和国内外同类研究对比.
2 结果与讨论 2.1 PM2.5化学组成由表 1可知, 采样期间PM2.5平均质量浓度为(81.6±36.4) μg·m-3, 远高于《环境空气质量标准》(GB 3095-2012)规定的年均二级质量浓度限值(35 μg·m-3). 如图 1(a)所示, PM2.5质量浓度在不同季节表现为冬季[(85.7±35.7) μg·m-3]和春季[(98.9±38.5) μg·m-3]高于夏季[(68.0±26.1) μg·m-3]和秋季[(74.5±37.6) μg·m-3]. 冬季除气象条件有利于污染物积累外(例如, 边界层高度降低, 大气稳定), 化石燃料燃烧排放急剧增加; 受相对湿度降低[采样期间平均RH:春(61.2%) < 秋(68.6%) < 冬(72.6%) < 夏(73.7%)]和沙尘暴影响, 春季扬尘对该地区PM2.5的贡献升高[27, 28]. 夏季边界层顶升高, 大气湍流运动强烈, 且降水量大, 污染物的清除速率加快. 由于夜间边界层高度的降低和大气稳定度的升高均可导致PM2.5积累, PM2.5在夜间的平均质量浓度[(84.0±37.7) μg·m-3]略高于白天[(79.2±35.4) μg·m-3], 但昼夜差异不显著(P=0.62).
|
|
表 1 采样期间PM2.5中化学组分的质量浓度及有机组分的吸光性质1) Table 1 Mass concentrations of PM2.5components and light-absorbing properties of bulk organics during the sampling period |

|
图 1 采样期间PM2.5中化学组分的质量浓度变化 Fig. 1 Mass concentration time-series of PM2.5 components during the sampling period |
无机离子和碳质组分是PM2.5的主要化学成分. NH4+、NO3-和SO42-的平均质量浓度分别为(4.99±3.32)、(12.0±10.3)和(8.83±4.08) μg·m-3(表 1). 如图 1(b)~1(d), 这3种离子的质量浓度均在冬季达到最大. 所有组分中, 仅有NO3-质量浓度存在显著的昼夜差异(P=0.047). PM2.5中NH4+、NO3-和SO42-的形成过程主要包括NOx和SO2的均相和非均相反应、反应产物(HNO3和H2SO4)和大气中碱性组分(例, NH3)的中和反应. 除边界层高度降低和大气稳定度升高外, 冬季和夜间低温且高湿的条件有利于NOx和SO2的二次反应过程; 而夏季和白天温度较高, 硝酸铵更容易分解挥发. SO42-的质量浓度在白天比夜间高约10%, 和白天更强的光化学反应有关. 如图 1(e)~1(g), K+、Mg2+和Ca2+质量浓度低, 其总质量浓度约为上述3种二次离子的12%. K+质量浓度没有明显的季节变化, 而Mg2+和Ca2+质量浓度在春季因扬尘贡献升高而增加.
表 1中碳质组分OC和EC的平均质量浓度分别为(8.26±3.51) μg·m-3和(2.87±1.10) μg·m-3. 其中OC的质量浓度高于2014~2018年南京地区PM2.5 OC的平均质量浓度(6.38 μg·m-3), 而EC则低于往年(3.12 μg·m-3)[29]. 南京地区PM2.5碳质组分的质量浓度低于北方城市, 例如北京(OC 12.4 μg·m-3, EC 3.7 μg·m-3)和天津(OC 12.0 μg·m-3, EC 3.1 μg·m-3), 高于东南沿海城市, 例如上海(OC 7.19 μg·m-3, EC 2.37 μg·m-3)和广州(OC 7.6 μg·m-3, EC 1.7 μg·m-3)[29, 30]. 另外, OC与EC的质量浓度比值(OC/EC=2.92±0.69)延续了2014~2018逐年升高的趋势[29], 表明二次贡献持续增加. 如图 1(h)~1(j)所示, OC、EC和WSOC的质量浓度变化趋势相似. 其中OC与EC有强相关性(r=0.83, P < 0.01), 并且在冬季达到最高(r=0.92, P < 0.01), 表明OC的质量浓度也和一次源排放密切相关. 因二次有机气溶胶(secondary organic aerosol, SOA)形成的影响, OC与EC的相关性在夏季最低(r=0.76, P < 0.01). 受一次和二次源排放的影响, WSOC与EC的相关性也存在类似的季节变化(冬季r=0.89, P < 0.01; 夏季r=0.60, P < 0.01). 另外, WSOC与K+在冬季未表现出强相关(r=0.52, P < 0.01), 接近于春季(r=0.58, P < 0.01)和秋季(r=0.53, P < 0.01), 稍高于夏季(r=0.40, P < 0.01). 笔者推测该地区PM2.5中WSOC的主要一次源为化石燃料燃烧, 而非生物质燃烧. 除气象因素外, WSOC的季节分布由化石燃料燃烧和二次形成主导.
2.2 BrC的吸光性质 2.2.1 光吸收系数(Abs365)如表 1所示, 采样期间Abs365, m的均值[(7.69±4.93)Mm-1]为Abs365, w[(3.22±2.18)Mm-1]的2.60倍, 与美国洛杉矶[31]、美国东南部[26]、北京[20]和西安[23]的研究结果相似(Abs365, m/Abs365, w 2.0~3.2). 因为和水相比, 甲醇能提取更多的有机物, 且MEOC中非水溶性组分的吸光作用比WSOC更强[17, 31]. 表 2比较了国内外不同地区PM2.5 Abs365, w和Abs365, m的差异. 本研究Abs365, w在不同季节的均值比该地区上一年低27.4%~36.1%[24], 主要和WSOC质量浓度下降有关. 尽管本研究中MEOC的平均质量浓度较上一年降低13.9%[24], Abs365, m的均值却未降低, 说明该地区具有强吸光作用的非水溶性MEOC排放并未减少.和西安类似, Abs365, w和Abs365, m在冬季的平均值[(5.41±3.06)Mm-1和(12.2±6.34)Mm-1]分别为夏季[(1.86±1.01)Mm-1和(4.57±2.86)Mm-1]的2.91和2.67倍. 美国城市地区PM2.5的Abs365, w(0.21~1.27 Mm-1)和Abs365, m(0.52~2.82 Mm-1)水平远低于我国城市地区, 而南京地区的Abs365, w和Abs365, m则远低于北京(10.2 Mm-1和26.2 Mm-1)和西安(5.0~25.2 Mm-1和8.3~46.3 Mm-1). 已有研究表明, 北京冬季BrC的吸光作用显著受生物质燃烧影响[20, 21], 而南京北郊MEOC、Abs365, m与K+质量浓度在冬季的弱相关(r为0.42和0.28, P < 0.01)说明生物质燃烧并不是该地区冬季BrC的主要一次源. 图 2(a)给出了Abs365, w、Abs365, m以及Abs365, m/Abs365, w比值随时间的变化. Abs365, w和Abs365, m分别与WSOC(r=0.72, P < 0.01)和MEOC(r=0.62, P=0.04)的质量浓度显著相关, 因此Abs365, w和Abs365, m的季节和区域性差异与PM2.5中有机组分质量浓度的时空分布密切相关. Abs365, w和Abs365, m在夜间的平均值[(3.47±2.36)Mm-1和(8.63±5.51)Mm-1]均高于白天[(2.97±1.97)Mm-1和(6.76±4.14)Mm-1]. 一方面, 夜间不利的气象条件有利于有机组分的积累; 另一方面PM2.5中的吸光性组分在白天因光解反应易发生损耗(“光漂白作用”)[32]. 与Abs365, w和Abs365, m的季节变化不同, Abs365, m/Abs365, w在夏季(2.62±1.06)高于冬季(2.39±0.59), 这是因为夏季WSOC受更多“光漂白作用”的影响导致对总吸光作用的贡献降低. Abs365, m/Abs365, w的年均值(2.60±0.92)远高于MEOC/WSOC(1.37±0.30), 表明MEOC中非水溶性组分的吸光作用更强, 在BrC的吸光作用中占主导地位.
|
|
表 2 南京与其它地区BrC吸光性质对比 Table 2 Comparisons of BrC absorption between Nanjing and other areas |

|
图 2 采样期间BrC吸光性质变化 Fig. 2 Temporal variations in light-absorbing properties of BrC during the sampling period |
如表 1所示, BrC的MAE365, w[(0.64±0.29)m2·g-1]和MAE365, m[(1.19±0.59)m2·g-1]均值远小于BC(11.3 m2·g-1)[31]. MAE365, w、MAE365, m和MAE365, m/MAE365, w的季节变化与Abs365相同, MAE365均表现为冬高[(1.05±0.25)m2·g-1和(1.86±0.44)m2·g-1]夏低[(0.40±0.14)m2·g-1和(0.79±0.47)m2·g-1], 而MAE365, m/MAE365, w为冬低(1.82±0.38)夏高[1.99±0.85, 图 2(b)]. 与Abs365, w类似, 本研究MAE365, w在不同季节的均值均低于该地区上一年水平, 而MAE365, m在春、秋和冬季均高于上一年(表 2)[24]. 主要可归结于吸光性WSOC质量浓度的显著降低, 而MEOC中主导BrC吸光作用的非水溶性组分却未减少. 另外, 本研究中夏季MAE365的数值远低于以生物质燃烧和人为排放为主导的洛杉矶(MAE365, w=0.71 m2·g-1和MAE365, m=1.58 m2·g-1)[31], 而更接近于美国东南部地区的夏季(0.29 m2·g-1和0.36 m2·g-1)[26]. 冬季MAE365, w和MAE365, m的均值与北京(1.22 m2·g-1和1.45 m2·g-1)处于同一水平(表 2)[20]. 表 1中水和甲醇对OC的提取效率(ηw和ηm)分别为(59.9±10.2)%和(80.2±8.51)%. 假设WSOC亦可溶于甲醇, MEOC-WSOC即为MEOC中的非水溶性部分. 由(Abs365, m- Abs365, w)/(CMEOC-CWSOC)计算得到该部分OC的MAE365高达(4.10±5.15)m2·g-1, 分别是MAE365, w和MAE365, m的6.0和2.9倍, 进一步证明MEOC中非水溶性组分的吸光强度远高于WSOC, 在BrC的吸光作用中占主导地位.
2.2.3 埃氏吸收指数(Å)Å常用于表征BrC吸光作用对波长的依赖性, Å越大表示吸光作用对波长变化越敏感.如表 1, ÅWSOC和ÅMEOC的平均值分别为7.09±1.12和6.17±1.46, 表明WSOC的吸光作用随波长的变化比MEOC更大. 本研究中ÅWSOC的季节(6.69±0.79~7.25±1.05)与昼夜(7.06±1.07~7.12±1.18)变化均不显著[P为0.05~0.95, 图 2(c)], 与美国东南部地区(6~9)[25, 26]、洛杉矶(7.6)[31]和北京(7.28)[20]的观测结果一致(表 2); ÅMEOC则体现出较明显的季节(冬季5.58±0.73, 夏季7.09±1.68)和空间变化(表 2). 实验室模拟研究表明, SOA的ÅWSOC对前体物类型和氧化程度的依赖性较低[33]. 因此笔者推测ÅWSOC在不同时间和空间范围内的一致性和WSOC中SOA的贡献有关, 而ÅMEOC的时空分布特征由MEOC中非水溶性组分的来源差异决定.
2.3 主成分分析利用主成分分析法(principal component analysis, PCA)定性识别PM2.5各丰量组分和有机组分吸光作用的主要来源. 表 3给出了在特征值大于1的条件下, 5个主因子在各组分上的负荷. 所有因子的方差累积贡献率达到91.7%, 较完整地解释了输入数据的方差. 因子1中NH4+和NO3-负荷最高, 该因子可代表硝酸铵的二次形成过程. Abs365, w和Abs365, m也在该因子中有最高的因子负荷. 由于NO3-的前体物NOx可通过氧化芳香类有机化合物生成具有强吸光作用的BrC[34], 铵盐或NH3介导作用下的液相反应也是BrC的重要二次来源[35], 笔者推测南京北郊大气中BrC主要来自NOx和NH3参与的各种二次反应过程. 因子2中的特征组分包括OC、EC和WSOC. 城市地区的人为活动是PM2.5中含碳物质的主要来源, 且采样点靠近交通干道, 交通排放的贡献更加突出, 所以因子2可代表和人为活动相关的一次排放源(例, 机动车排放). Abs365, w与Abs365, m在因子2中负荷仅次于因子1, 说明人为活动相关的一次排放也是该地区BrC的重要来源. Mg2+和Ca2+在因子3中负荷最高. 南京市位于内陆地区, PM2.5中的Mg2+主要来自地壳而非海洋, 而Ca2+是扬尘的重要标志物[36]. 因此, 因子3体现了采样点附近建筑和交通扬尘的贡献. 值得注意的是, Abs365, m在因子3中的负荷远低于Abs365, w, 潜在原因为扬尘中的HULIS具有较强吸光作用, 是WSOC的重要组成部分(9%~72%)[37]. SO42-主要由工业排放的SO2通过液相氧化或非均相过程产生, 从因子4中各组分负荷分布可知, 该因子代表工业排放相关的二次硫酸盐生成. 因子5中K+的负荷最大, 且PM2.5中K+被认为是生物质燃烧的示踪物, 但Abs365, w和Abs365, m的负荷仅有0.23和0.32, 说明因子5所代表的生物质燃烧不是该地区BrC的主要来源.
|
|
表 3 基于PM2.5组成和BrC吸光作用的主成分分析结果1) Table 3 PCA results of PM2.5 components and BrC absorption |
3 结论
(1) 受气象条件、源排放和“光漂白作用”的影响, 南京北郊PM2.5中WSOC和MEOC的吸光作用均表现为冬高夏低, 夜高昼低的时间变化特征.
(2) 通过比较WSOC和MEOC的质量浓度和吸光性质, 得出MEOC中非水溶性组分的吸光强度远高于WSOC, 在BrC的吸光作用中占主导地位.
(3) 和MEOC相比, WSOC的吸光作用对波长变化更加敏感, 而MEOC吸光作用对波长的依赖性随时间变化更显著, 这和MEOC中非水溶性组分的来源变化有关.
(4) 相关性和主成分分析结果均表明, 生物质燃烧不是该地区PM2.5中BrC的主要一次来源. 该地区BrC的吸光作用主要和NOx相关的光氧化反应、铵盐或NH3参与的气相或非均相反应以及人为活动相关的一次排放源(例, 机动车排放)有关.
| [1] | Peng J F, Hu M, Guo S, et al. Markedly enhanced absorption and direct radiative forcing of black carbon under polluted urban environments[J]. Proceedings of the National Academy of Sciences of the United States of America, 2016, 113(16): 4266-4271. DOI:10.1073/pnas.1602310113 |
| [2] | Ramanathan V, Carmichael G. Global and regional climate changes due to black carbon[J]. Nature Geoscience, 2008, 1(4): 221-227. DOI:10.1038/ngeo156 |
| [3] |
张小曳. 中国大气气溶胶及其气候效应的研究[J]. 地球科学进展, 2007, 22(1): 12-16. Zhang X Y. Aerosol over China and their climate effect[J]. Advances in Earth Science, 2007, 22(1): 12-16. DOI:10.3321/j.issn:1001-8166.2007.01.002 |
| [4] | Zhang X Y, Wang Y Q, Niu T, et al. Atmospheric aerosol compositions in China: spatial/temporal variability, chemical signature, regional haze distribution and comparisons with global aerosols[J]. Atmospheric Chemistry and Physics, 2012, 12(2): 779-799. DOI:10.5194/acp-12-779-2012 |
| [5] | Bond T C, Streets D G, Yarber K F, et al. A technology-based global inventory of black and organic carbon emissions from combustion[J]. Journal of Geophysical Research: Atmospheres, 2004, 109(D14). DOI:10.1029/2003JD003697 |
| [6] | Myhre G, Samset B H, Schulz M, et al. Radiative forcing of the direct aerosol effect from AeroCom Phase Ⅱ simulations[J]. Atmospheric Chemistry and Physics, 2013, 13(4): 1853-1877. DOI:10.5194/acp-13-1853-2013 |
| [7] | Saleh R, Hennigan C J, McMeeking G R, et al. Absorptivity of brown carbon in fresh and photo-chemically aged biomass-burning emissions[J]. Atmospheric Chemistry and Physics, 2013, 13(15): 7683-7693. DOI:10.5194/acp-13-7683-2013 |
| [8] | Xie M J, Hays M D, Holder A L. Light-absorbing organic carbon from prescribed and laboratory biomass burning and gasoline vehicle emissions[J]. Scientific Reports, 2017, 7(1). DOI:10.1038/s41598-017-06981-8 |
| [9] | Iinuma Y, Böge O, Gräfe R, et al. Methyl-nitrocatechols: atmospheric tracer compounds for biomass burning secondary organic aerosols[J]. Environmental Science & Technology, 2010, 44(22): 8453-8459. |
| [10] | Liu J M, Lin P, Laskin A, et al. Optical properties and aging of light-absorbing secondary organic aerosol[J]. Atmospheric Chemistry and Physics, 2016, 16(19): 12815-12827. DOI:10.5194/acp-16-12815-2016 |
| [11] | Laskin A, Laskin J, Nizkorodov S A. Chemistry of atmospheric brown carbon[J]. Chemical Reviews, 2015, 115(10): 4335-4382. DOI:10.1021/cr5006167 |
| [12] | Feng Y, Ramanathan V, Kotamarthi V R. Brown carbon: a significant atmospheric absorber of solar radiation?[J]. Atmospheric Chemistry and Physics, 2013, 13(17): 8607-8621. DOI:10.5194/acp-13-8607-2013 |
| [13] | Zhang Y Z, Forrister H, Liu J M, et al. Top-of-atmosphere radiative forcing affected by brown carbon in the upper troposphere[J]. Nature Geoscience, 2017, 10(7): 486-489. DOI:10.1038/ngeo2960 |
| [14] | Chung C E, Ramanathan V, Decremer D. Observationally constrained estimates of carbonaceous aerosol radiative forcing[J]. Proceedings of the National Academy of Sciences of the United States of America, 2012, 109(29): 11624-11629. DOI:10.1073/pnas.1203707109 |
| [15] | Yang M, Howell S G, Zhuang J, et al. Attribution of aerosol light absorption to black carbon, brown carbon, and dust in China-interpretations of atmospheric measurements during EAST-AIRE[J]. Atmospheric Chemistry and Physics, 2009, 9(6): 2035-2050. DOI:10.5194/acp-9-2035-2009 |
| [16] | Lu Z, Zhang Q, Streets D G. Sulfur dioxide and primary carbonaceous aerosol emissions in China and India, 1996-2010[J]. Atmospheric Chemistry and Physics, 2011, 11(18): 9839-9864. DOI:10.5194/acp-11-9839-2011 |
| [17] | Liu J, Bergin M, Guo H, et al. Size-resolved measurements of brown carbon in water and methanol extracts and estimates of their contribution to ambient fine-particle light absorption[J]. Atmospheric Chemistry and Physics, 2013, 13(24): 12389-12404. DOI:10.5194/acp-13-12389-2013 |
| [18] | Zhang X L, Lin Y H, Surratt J D, et al. Light-absorbing soluble organic aerosol in Los Angeles and Atlanta: a contrast in secondary organic aerosol[J]. Geophysical Research Letters, 2011, 38(21). DOI:10.1029/2011GL049385 |
| [19] | Xie M J, Chen X, Hays M D, et al. Light absorption of secondary organic aerosol: composition and contribution of nitroaromatic compounds[J]. Environmental Science & Technology, 2017, 51(20): 11607-11616. |
| [20] | Cheng Y, He K B, Du Z Y, et al. The characteristics of brown carbon aerosol during winter in Beijing[J]. Atmospheric Environment, 2016, 127: 355-364. DOI:10.1016/j.atmosenv.2015.12.035 |
| [21] | Li X R, Yang Y, Liu S Q, et al. Light absorption properties of brown carbon (BrC) in autumn and winter in Beijing: composition, formation and contribution of nitrated aromatic compounds[J]. Atmospheric Environment, 2020, 223. DOI:10.1016/j.atmosenv.2020.117289 |
| [22] | Yuan J F, Huang X F, Cao L M, et al. Light absorption of brown carbon aerosol in the PRD region of China[J]. Atmospheric Chemistry and Physics, 2016, 16(3): 1433-1443. DOI:10.5194/acp-16-1433-2016 |
| [23] | Huang R J, Yang L, Cao J J, et al. Brown carbon aerosol in urban Xi'an, northwest China: the composition and light absorption properties[J]. Environmental Science & Technology, 2018, 52(12): 6825-6833. |
| [24] | Xie X C, Chen Y F, Nie D Y, et al. Light-absorbing and fluorescent properties of atmospheric brown carbon: a case study in Nanjing, China[J]. Chemosphere, 2020, 251. DOI:10.1016/j.chemosphere.2020.126350 |
| [25] | Hecobian A, Zhang X, Zheng M, et al. Water-Soluble Organic Aerosol material and the light-absorption characteristics of aqueous extracts measured over the southeastern United States[J]. Atmospheric Chemistry and Physics, 2010, 10(13): 5965-5977. DOI:10.5194/acp-10-5965-2010 |
| [26] | Xie M J, Chen X, Holder A L, et al. Light absorption of organic carbon and its sources at a southeastern U.S. location in summer[J]. Environmental Pollution, 2019, 244: 38-46. DOI:10.1016/j.envpol.2018.09.125 |
| [27] | Yu Y Y, He S Y, Wu X L, et al. PM2.5 elements at an urban site in Yangtze River Delta, China: high time-resolved measurement and the application in source apportionment[J]. Environmental Pollution, 2019, 253: 1089-1099. DOI:10.1016/j.envpol.2019.07.096 |
| [28] | Yu Y Y, Ding F, Mu Y F, et al. High time-resolved PM2.5 composition and sources at an urban site in Yangtze River Delta, China after the implementation of the APPCAP[J]. Chemosphere, 2020, 261. DOI:10.1016/j.chemosphere.2020.127746 |
| [29] |
丁峰, 朱志锋, 陆晓波, 等. 2014-2018年南京PM2.5中碳组分污染特征分析[J]. 中国环境监测, 2020, 36(2): 165-172. Ding F, Zhu Z F, Lu X B, et al. Characteristics analysis of carbon components in PM2.5 in Nanjing from 2014 to 2018[J]. Environmental Monitoring in China, 2020, 36(2): 165-172. |
| [30] | Wang J Z, Ho S S H, Ma S X, et al. Characterization of PM2.5 in Guangzhou, China: uses of organic markers for supporting source apportionment[J]. Science of the Total Environment, 2016, 550: 961-971. DOI:10.1016/j.scitotenv.2016.01.138 |
| [31] | Zhang X L, Lin Y H, Surratt J D, et al. Sources, composition and absorption Ångström exponent of light-absorbing organic components in aerosol extracts from the Los Angeles Basin[J]. Environmental Science & Technology, 2013, 47(8): 3685-3693. |
| [32] | Fasnacht M P, Blough N V. Mechanisms of the aqueous photodegradation of polycyclic aromatic hydrocarbons[J]. Environmental Science & Technology, 2003, 37(24): 5767-5772. |
| [33] | Lambe A T, Cappa C D, Massoli P, et al. Relationship between oxidation level and optical properties of secondary organic aerosol[J]. Environmental Science & Technology, 2013, 47(12): 6349-6357. |
| [34] | Jacobson M Z. Isolating nitrated and aromatic aerosols and nitrated aromatic gases as sources of ultraviolet light absorption[J]. Journal of Geophysical Research: Atmospheres, 1999, 104(D3): 3527-3542. DOI:10.1029/1998JD100054 |
| [35] | Bones D L, Henricksen D K, Mang S A, et al. Appearance of strong absorbers and fluorophores in limonene-O3 secondary organic aerosol due to NH4+-mediated chemical aging over long time scales[J]. Journal of Geophysical Research: Atmospheres, 2010, 115(D5). DOI:10.1029/2009JD012864 |
| [36] | Shen Z X, Sun J, Cao J J, et al. Chemical profiles of urban fugitive dust PM2.5 samples in Northern Chinese cities[J]. Science of the Total Environment, 2016, 569-570: 619-626. DOI:10.1016/j.scitotenv.2016.06.156 |
| [37] |
项萍, 谭吉华, 马永亮, 等. 大气颗粒物中类腐殖酸的研究进展[J]. 环境化学, 2015, 34(3): 401-409. Xiang P, Tan J H, Ma Y L, et al. Research progress of humic-like substances (HULIS) in atmospheric particles[J]. Environmental Chemistry, 2015, 34(3): 401-409. |