 2020, Vol. 41
2020, Vol. 41



2. 成都理工大学国家环境保护水土污染协同控制与联合修复重点实验室, 成都 610059;
3. 中国环境科学研究院环境基准与风险评估国家重点实验室, 北京 100012
2. State Environmental Protection Key Laboratory of Synergetic Control and Joint Remediation for Soil&Water Pollution, Chengdu University of Technology, Chengdu 610059, China;
3. State Key Laboratory of Environmental Criteria and Risk Assessment, China Academy of Environmental Sciences, Beijing 100012, China
对-硝基苯酚(PNP)具有高毒性、致癌性和生物蓄积性, 通常从合成染料、石化、增塑剂、农药和制药等行业释放到环境中[1, 2].PNP存在范围广、毒性大且降解速度慢的特点导致了严重的环境问题.对-硝基苯酚通常使用高级氧化技术去除, 其中, 过渡金属催化剂活化过一硫酸盐(PMS)产生活性自由基的方式是研究热点之一, 特别是双金属催化剂具备的独特电子、光学和催化特性引起了更多学者的关注[3~6].双金属催化剂的结构, 例如异质结构、金属间结构、合金结构和核壳结构, 会对其物理化学性质产生巨大影响.其中, 核壳结构由于其许多优点在催化领域具有巨大的潜力[7].例如, 双金属催化剂的核与壳之间提供的巨大反应界面能够提高其催化活性[8]; 金属核充当电子源并发挥还原作用, 壳通过静电相互作用和表面络合促进污染物的吸附, 并允许电子从金属核中通过[9]; 壳出色地耐用性可以保护核, 阻碍催化剂颗粒团聚[10, 11].
磁铁矿(Fe3 O4)由于比表面积大、活性位点多和强磁性的特点在工程应用中显示出许多优势, 常与其他材料进行复合[12, 13].过渡金属铜由于其高效的活化PMS性能备受关注, 但是使用纳米零价铜(Cu0)活化PMS的研究较少.已有的研究中, Cu0/PMS体系通常表现出严重的pH依赖性, 在酸性条件下凭借Cu0表面腐蚀释放出的铜离子, 对PMS进行均相活化, 界面反应程度很低, 存在二次污染等缺陷[14, 15].由于Cu+还原Fe3+在热力学上是可行的, 可以认为铁铜双金属间应该存在协同作用可以增强催化剂的活性.在Nie等[16]的研究中, Cu0-Fe3 O4微米级催化剂表现出了Fe-Cu双金属协同作用, 但其在pH值大于7后仍表现出很差的降解性能.Pham等[17, 18]的研究中则通过硼氢化钠还原的方法成功制备了Cu0@ Fe3 O4催化剂, 并用其活化H2 O2降解土霉素, 在pH值为9时仍能达到接近65%的降解率.这说明Fe-Cu双金属协同和核壳结构改变pH适应范围理论上是可行的.
为了改善Cu0/PMS体系适应pH范围窄的缺陷, 本研究通过共沉淀法制备了纳米Cu0@ Fe3 O4复合催化剂, 利用复合催化剂的核壳结构改变其活化PMS的方式, 扩大其pH适应范围.以PNP为特征污染物, 研究了Cu0@ Fe3 O4活化PMS体系的反应机制和PNP的协同降解途径.
1 材料与方法 1.1 实验材料硫酸亚铁、硫酸铁、硫酸、磷酸氢二钾、磷酸二氢钾、抗坏血酸和四硼酸钠(均为分析纯, 成都科龙化工试剂厂); 无水乙醇、叔丁醇和氢氧化钠(均为分析纯, 成都科隆化学品有限公司); PMS(优级纯, 上海阿拉丁生化股份有限公司); 纳米铜粉(10~30 nm)、PNP、碘化钾和新亚铜试剂(上海阿拉丁生化股份有限公司).
1.2 纳米Cu0@ Fe3 O4复合催化剂制备Cu0@ Fe3 O4纳米磁性核壳材料通过共沉淀法制备, 如图 1所示.主要原理是将纳米铜粉(10~30 nm)置入Fe3 O4合成体系中, 铜为Fe3 O4生成提供附着点从而形成核壳结构, 具体操作如下:使用100 mL去离子水将Fe2(SO4)3(0.6 g), FeSO4·7H2 O(0.417 g)溶解在三颈圆底烧瓶中; 在N2、70℃的条件下持续搅拌0.5 h后, 用400 mL去离子水稀释混合溶液; 将0.285 g纳米铜粉(铜铁摩尔比=1:1)加入100 mL NaOH(1 g)溶液中, 细胞破碎40 min, 使纳米铜粉均匀分散在NaOH溶液中.向铁混合溶液中快速加入NaOH-Cu0混合溶液, 形成黑棕色沉淀; 0.5 h后, 固体沉淀物通过外部磁场分离, 并用去离子水洗涤几次后使用真空干燥箱在60℃干燥12 h.

|
图 1 共沉淀法制备纳米Cu0@ Fe3 O4复合催化剂流程 Fig. 1 Schematic diagram of nano-Cu0@ Fe3 O4 composite catalyst prepared using co-precipitation method |
制备所得纳米Cu0@ Fe3 O4复合催化剂通过场发射电镜(SEM, Sigma300, 德国Zeiss)和高倍透射电镜(HRTEM, JEM-2100, 日本电子株式会社)表征催化剂的表面形态和结构, 通过X-射线粉末衍射仪(XRD, D8 Advance, 德国布鲁克公司)验证催化剂的元素和晶体结构, 通过X射线粉末衍射仪(XPS, K-Alpha, 美国赛默飞公司)表征了催化剂反应前后的元素价态变化.
1.4 PNP降解批实验所有降解实验均在250 mL锥形瓶中进行, 该瓶中装有100 mL 5 mg·L-1 PNP溶液, 置于(25±2)℃的恒温水浴振荡箱.首先将催化剂与锥形瓶中的PNP溶液混合, 超声5 min后加入一定量的PMS以引发降解过程(吸附实验表明催化剂对PNP无吸附性, 故批实验未进行吸附实验).在实验过程中恒温水浴振荡箱的转速为150 r·min-1.在每个采样点用注射器采集0.5 mL样品, 并通过添加等体积的乙醇淬灭, 使用0.22 μm滤头过滤后加入四硼酸钠缓冲溶液调节pH至碱性.使用酶标仪在405 nm处测试PNP吸光度.在猝灭实验中, 将乙醇(EtOH)和叔丁醇(TBA)作为硫酸根自由基(SO4-·)和羟基自由基(HO·)的捕获剂.
所有实验均作3个平行样, 图中的所有数据均为平均值, 误差线代表平均值的标准偏差.
1.5 检测分析方法 1.5.1 PNP检测PNP在酸性条件下特征峰在300 nm左右, 不显色; 碱性条件下特征峰可在405 nm左右测得, 显黄色.为方便测定, 样品取出后加入等体积的乙醇进行猝灭, 然后使用四硼酸钠缓冲液(pH=9.18)调节pH至碱性后取出0.25 mL溶液使用酶标仪在405 nm处进行测定, 即得PNP的最大吸光度.为保证测定结果的可比性, 每个样品加入的四硼酸钠缓冲液体积一致, 对不同浓度PNP的检测结果稳定.在不同初始pH影响实验中, 四硼酸钠缓冲液能够调节测试样品至碱性并充分显色, 不同降解时间节点的最大吸收波长不发生偏移, 保证测定结果可靠.酶标仪作为多通道分光光度计使用.
1.5.2 PMS浓度检测使用酶标仪通过碘量法测试PMS的浓度.将5 g碳酸氢钠, 50 g碘化钾和50 mL去离子水(电阻率18.25 MΩ·cm)混合用于配制储备溶液.将1 mL过滤的PMS样品加入到9 mL储备溶液中.充分显色20 min后, 使用酶标仪在354 nm处检测PMS的浓度.
1.5.3 TCu、TCu+以及铁离子浓度检测(1) 总亚铜离子浓度(TCu+)将1 mL过滤的样品, 0.2 mL 0.5 mol·L-1磷酸-磷酸盐缓冲液, 0.2 mL 10 mmol·L-1新亚铜试剂(NP:C14H12N2)和8.6 mL超纯水(电阻率18.25 MΩ·cm)混合以产生Cu+-NP复合物.通过酶标仪在454 nm处检测TCu+的浓度.
(2) 总铜浓度(TCu)添加抗坏血酸将Cu2+还原成Cu+进行测定.将1 mL过滤的样品, 0.2 mL 0.5 mmol·L-1磷酸-磷酸盐缓冲液, 0.2 mL 10 mmol·L-1 NP, 0.2 mL 0.1 mmol·L-1抗坏血酸和8.4 mL超纯水(电阻率18.25 MΩ·cm)混合以产生Cu+-NP复合物.通过酶标仪在454 nm处检测TCu的浓度.
(3) 铁离子浓度通过火焰原子吸收实验测定(过滤后)反应结束后样品中铁离子浓度.
2 结果与讨论 2.1 纳米Cu0@ Fe3 O4复合催化剂的表征采用FESEM观察了纳米Cu0@ Fe3 O4反应前后的表面形貌变化, 如图 2所示.由于催化剂制备时添加的Cu0可能存在部分未分散开的团聚体, 从而使得极少量观测到的催化剂粒径较大.为了更清晰地查看反应前后催化剂的表面形貌变化, 此处展示的是大颗粒催化剂的FESEM图.图 2(a)和2(b)展示了催化剂在未发生反应时的表面形貌, 主要呈现球状, 表面为Fe3 O4壳层, 未见Cu0.在图 2(c)和2(d)反应后的纳米Cu0@ Fe3 O4复合物FESEM图中可发现, 球状的催化剂表面壳层出现轻微的剥离, 并显现出部分Cu0的表面形貌.

|
(a)和(b)反应前; (c)和(d)反应后 图 2 反应前和反应后纳米Cu0@ Fe3 O4复合物的FESEM图像 Fig. 2 FESEM images of unreacted and reacted nano-Cu0@ Fe3 O4 composite |
图 3(a)和3(c)可以看出, 纳米Cu0@ Fe3 O4复合物颗粒直径约为(10.28±2.13)nm, 存在极少的团聚大颗粒(d≈100 nm).图 3(b)中纳米粒子存在0.189 nm和0.262 nm的晶格条纹间距, 分别与Cu0的(200)晶面以及Fe3 O4的(311)晶面相匹配.在图 3(b)可以看出, Cu0在颗粒内部, 为催化剂的核, 而Fe3 O4包覆在Cu0的表面, 为催化剂的壳.催化剂的HRTEM表征显示出了纳米Cu0@ Fe3 O4复合物的核壳结构.

|
(a)纳米Cu0@ Fe3 O4复合物的HRTEM图像; (b)显示纳米Cu0@ Fe3 O4复合物核壳结构的HRTEM图像; (c)纳米Cu0@ Fe3 O4复合物的粒径分布(不少于100个颗粒计数) 图 3 纳米Cu0@ Fe3 O4复合物的HRTEM图像和粒径分布 Fig. 3 HRTEM image and particle size distribution diagram of nano-Cu0@ Fe3 O4 composite |
随后, 将Fe3 O4、Cu0、Cu0@ Fe3 O4和反应后的Cu0@ Fe3 O4进行了XRD光谱表征, 如图 4(a)所示.在Fe3 O4的XRD光谱图中, 在2θ为35.5°、38.7°、42.6°和61.4°处出现峰值, 与标准四氧化三铁的XRD标准卡片(JCPD No. 79-0417)相对应.在Cu0的XRD光谱图中, 在2θ为43.3°、50.3°和74.0°处出现峰值, 与铜纳米颗粒的XRD标准卡片(JCPD No.85-1326)相对应.在Cu0@ Fe3 O4的XRD光谱图中, 可以看出只存在Fe3 O4的特征峰, 而无Cu0的特征峰, 证实了Cu0@ Fe3 O4的核壳结构, 只检测到作为壳的Fe3 O4特征峰.在反应后的Cu0@ Fe3 O4 XRD光谱图中, 则同时出现了Fe3 O4和Cu0的特征峰, 可能是因为反应后部分催化剂表层的Fe3 O4壳被剥离, 导致Cu0暴露后被检测到特征峰, 与FESEM表征现象一致.

|
图 4 纳米Cu0@ Fe3 O4复合物的XRD光谱图和磁滞回线 Fig. 4 XRD spectrum and hysteresis loop of nano-Cu0@ Fe3 O4 composite |
图 4(b)所示为反应前后Cu0@ Fe3 O4催化剂的磁滞曲线, 从中可以看出反应后Cu0@ Fe3 O4的磁感应强度较反应前有一定的减弱, 但总体来说两者均具有较好的磁性, 能够通过外加磁场进行固液分离, 具备磁性回收和重复利用的能力.
2.2 纳米Cu0@ Fe3 O4的催化性能图 5(a)显示了150 mg·L-1的纯Fe3 O4、Cu0和Cu0@ Fe3 O4这3种催化剂对5 mg·L-1 PNP的吸附能力, 分别为10%、6%和4%, 其中Cu0和Cu0@ Fe3 O4对PNP的吸附能力几乎可以被忽略.如图 5(b), PMS剂量为5 mmol·L-1时, 相同条件的Fe3 O4/PMS、Cu0/PMS和Cu0@ Fe3 O4/PMS体系对PNP的降解率分别为8%、53%和90%. Cu0@ Fe3 O4/PMS体系中PNP的降解率远高于Fe3 O4/PMS、Cu0/PMS体系以及两体系的降解率总和(61%).以上结果证明纳米Cu0@ Fe3 O4催化剂具有优异的催化性能, 并且Cu0@ Fe3 O4/PMS体系中除了纯Fe3 O4和Cu0的贡献, 还存在Fe-Cu双金属协同作用.同时, 图 5(c)展示了不同反应体系的PMS消耗率, Fe3 O4/PMS、Cu0/PMS和Cu0@ Fe3 O4/PMS体系的PMS消耗率分别为36%, 69%和71%.纯Fe3 O4对PMS的消耗率最低, 与其对PNP的降解率最低相对应, 说明纯Fe3 O4很难有效活化PMS, 从而达到对污染物的高降解率. Cu0和Cu0@ Fe3 O4对PMS的相对消耗量比较接近, 但结合两种催化剂活化PMS降解PNP的速率和降解率, 不难推测Cu0@ Fe3 O4分解PMS的速率高于纯Cu0.

|
[催化剂]=150mg·L-1; [PMS]=0.5 mmol·L-1; [PNP]=5 mg·L-1; pH0=5.65; 25℃ 图 5 不同反应体系对PNP的吸附率和降解率以及反应过程中PMS的消耗量和铜离子的浸出浓度 Fig. 5 Adsorption and degradation rate of PNP in different reaction systems, the consumption of PMS and the leaching concentration of copper ions during the reaction |
为了验证Cu0@ Fe3 O4的核壳结构是否能够减少铜离子的浸出, 检测了Cu0@ Fe3 O4、Cu0+Fe3 O4和Cu0这3种催化剂活化PMS降解PNP过程中TCu和TCu+的浸出[图 5(d)].在30 min内, 其浸出的TCu浓度分别为1.9、14.0和23.18 mg·L-1, TCu+浓度分别为0.09、1.01和0.97 mg·L-1.其中TCu浓度占催化剂中铜总量的4.42%、32.56%和54.9%, 即浸出比例:Cu0@ Fe3 O4<Cu0+Fe3 O4<Cu0.另外还检测了反应后浸出铁离子的浓度, 结果表明铁的浸出浓度低于火焰原子分光光度计的检出限值(0.1mg·L-1).以上结果可以明显看出, Cu0@ Fe3 O4/PMS体系中的铜离子浸出远低于Cu0+Fe3 O4/PMS和Cu0/PMS体系, 铁离子浸出液极低, 几乎可以忽略, 是非均相界面反应.
2.3 降解过程主要影响因素 2.3.1 催化剂剂量不同Cu0@ Fe3 O4剂量对活化PMS降解PNP的影响如图 6(a)所示, 当Cu0@ Fe3 O4剂量为150 mg·L-1时, PNP的降解率为79%.随着Cu0@ Fe3 O4剂量的增加, PNP的降解速率和效率均有所增加, 主要是因为催化剂剂量的增加为反应体系提供了更多的活化位点.当催化剂剂量增加到200 mg·L-1和250 mg·L-1时, PNP降解效率分别为96%和97%, 差别不大, 因此出于节约成本的角度, 后续实验均为200 mg·L-1的催化剂投加量.

|
[催化剂]=200mg·L-1 [图(b)~(d)]; [PMS]=0.5mmol·L-1[图(a)、(c)和(d)]; [PNP]=5mg·L-1; [共存离子]=10mmol·L-1[图(c)和(d)]; pH0=5.65; 25℃ 图 6 不同催化剂剂量、PMS剂量和阴阳离子对PNP降解效率的影响 Fig. 6 Effects of different catalyst dosage, PMS dosage, and coexisting ions on PNP degradation efficiency |
PMS是一种活性氧前体, 因此PMS剂量会对Cu0@ Fe3 O4/PMS体系中PNP的降解有显著的影响.如图 6(b)所示, 当催化剂剂量固定为200 mg·L-1, PMS剂量从0.2 mmol·L-1增加到0.5 mmol·L-1时, 30 min内PNP的降解率从68%增加到了87%.在其他PMS/非均相催化体系中, 增加PMS浓度后也观察到了类似的效果[16].而当PMS剂量增加到0.6 mmol·L-1时, 降解效率有所下降, 这是由于过量的PMS对硫酸根自由基的猝灭作用[19].
2.3.3 共存离子由于在实际水体中PNP可能与一些无机离子共存, 本实验研究了无机离子碳酸氢根离子(HCO3-)、氯离子(Cl-)、磷酸氢根(H2PO42-)和硫酸根(SO42-)对PNP在Cu0@ Fe3 O4/PMS体系中降解的影响[图 6(c)].结果表明, H2PO42-和SO42-对PNP的降解存在抑制作用, 抑制程度为H2PO42->SO42-, 而HCO3-和Cl-对其降解存在促进作用, 促进程度为HCO3->Cl-. H2PO42-的抑制作用是因为H2PO42-在水性介质中可能与铁形成复合物, Fe2+不能自由地与溶液中的PMS反应[20].SO42-的抑制作用可能是由于离子强度高, 导致过硫酸根离子的分解较慢[21, 22]. HCO3-可与PMS发生反应产生过碳酸化合物, 当HCO3-浓度较低时, 其可与体系中过量的PMS反应从而表现出更强的氧化能力, 促进了PNP的降解[21]. Cl-对PNP降解反应性能的积极作用可能是由于通过一系列链反应生成了额外的自由基, 例如Cl·和Cl2-· [式(1)~(6)][23]:

|
(1) |
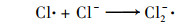
|
(2) |

|
(3) |
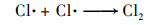
|
(4) |
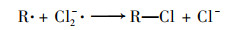
|
(5) |
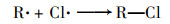
|
(6) |
图 6(d)展示了金属阳离子Ca2+、Mg2+、Zn2+对PNP降解的影响. 10 mmol·L-1的Ca2+和Mg2+对PNP降解的影响很微弱, 可以忽略不计.Zn2+对PNP降解具有抑制作用, 可能是因为Cu0@ Fe3 O4表面的活性位具有竞争性吸附作用[24, 25].
2.3.4 溶液初始pH为了探究Cu0@ Fe3 O4的核壳结构是否能够增强Cu0/PMS体系在碱性条件下对污染物的降解能力, 研究了初始pH值对Cu0@ Fe3 O4/PMS和Cu0/PMS两个体系的影响.图 7(a)显示了Cu0@ Fe3 O4/PMS体系在不同初始pH值下对PNP的降解能力, 结果表明PNP在5.56~9.06的pH范围内都能较好地被降解, 在pH为3.01时降解效率较低, 约46%, 可能是由于酸催化PMS分解, 消耗PMS的同时却不会产生自由基[26].值得注意的是, 在这两个体系中, pH值为11时PNP都能够被迅速降解70% ~80%, 并保持稳定, 这种现象并不代表催化剂的作用, 而是PMS能够在极碱性条件下被迅速活化, 从而产生活性自由基降解污染物[27].图 7(b)中初始pH值对Cu0/PMS体系的影响与其他研究报道的结果一致, 在3.01~9.05的pH范围内, 随着pH值的升高, 纳米零价铜的腐蚀程度降低, 从而PNP的降解率下降.这主要是因为纳米零价铜的零电位点一般在5.17附近, 高于其pHPZC时具有负电荷, 而在pH值低于其pHPZC值时具有正电荷, pH升高后可能会产生大量的负电荷, 从而抑制铜颗粒表面与HSO5-之间的静电相互作用, 抑制HSO5-加速Cu0腐蚀的能力[15].

|
[催化剂]=200mg·L-1; [PMS]=0.5mmol·L-1; [PNP]=5mg·L-1; 25℃ 图 7 初始pH值对Cu0@ Fe3 O4/PMS体系和Cu0/PMS体系的影响 Fig. 7 Effect of initial pH on Cu0@ Fe3 O4/PMS system and Cu0/PMS system |
对比初始pH值对两个反应体系的影响, 可以明显发现两者规律的不同, 说明Cu0@ Fe3 O4的核壳结构改善了单纯Cu0/PMS体系在碱性条件下降解性能较差的缺陷. Cu0@ Fe3 O4/PMS体系在不同初始pH环境中的表现更多地符合非均相活化和界面反应的规律, 改变了传统Cu0在反应体系中依赖腐蚀释放铜离子从而活化PMS的均相活化方式.
2.4 降解反应机制 2.4.1 自由基淬灭反应机制为了进一步对Cu0@ Fe3 O4/PMS体系降解PNP的机制进行探究, 使用乙醇(EtOH)和叔丁醇(TBA)作为自由基清除剂对体系产生的SO4-·和HO·进行了鉴定.据报道, 乙醇与HO·、SO4-·的二级反应速率都较高, 可以同时猝灭HO·和SO4-·;而叔丁醇与HO·的二级反应速率常数比其与SO4-·的快3个数量级, 可以选择性猝灭体系HO·[28].
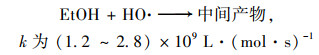
|
(7) |
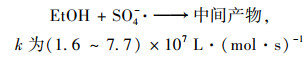
|
(8) |
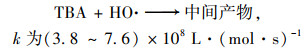
|
(9) |
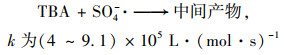
|
(10) |
在猝灭实验中, 添加的自由基清除剂与PMS浓度的比值从0~3 000不等, 随着添加剂量的增加, PNP的抑制效果也更为明显, 比值上升至3 000时乙醇和叔丁醇对PNP的抑制作用都基本达到稳定. Zhang等[19]将叔丁醇的猝灭效率等同于HO·的贡献, 而乙醇的猝灭效率则视为SO4-·和HO·的共同贡献. Du等[29]也因为乙醇和叔丁醇对双酚A的抑制率分别为23.1%和11.9%, SO4-·的贡献度更高.可使用公式(11)和(12)分别计算HO·和SO4-·对反应体系的贡献率[30].如图 8(a)所示, [TBA:PMS]=3 000:1时, PNP在30 min内的降解率从87%降低至35%, 说明体系中存在HO·, 按照式(11)计算得到HO·的贡献度为60%.在图 8(b)中, [EtOH:PMS]=3 000:1时, PNP在30 min内的降解率从87%降低至5%, 达到94%的抑制率, 接近完全抑制, 一般认为体系中只存在HO·和SO4-·, 按照式(12)计算得到SO4-·的贡献度为34%.这说明在Cu0@ Fe3 O4/PMS体系中, 同时存在HO·和SO4-·, 并且HO·是体系的主导自由基, 对PNP的降解起到重要的主导作用.
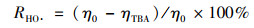
|
(11) |
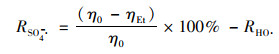
|
(12) |

|
[催化剂]=200mg·L-1; [PMS]=0.5mmol·L-1; [PNP]=5mg·L-1; pH0=5.65; 25℃ 图 8 叔丁醇和乙醇作为分子探针对Cu0@ Fe3 O4/PMS体系降解PNP的影响 Fig. 8 Effect of tert-butanol and ethanol as molecular probes on the degradation of PNP by Cu0@ Fe3 O4/PMS system |
式中, R为自由基贡献率(%); η0为不加猝灭剂时PNP在30 min内的降解率(%); ηEt为加入乙醇时PNP在30 min内的降解率(%); ηTBA为加入叔丁醇时PNP在30 min内的降解率(%).
2.4.2 体系中Fe-Cu协同机制为研究铁铜双金属的协同机制, 使用XPS表征了反应前后Cu0@ Fe3 O4中铁铜价态的变化.图 9(a)中, 检测到的化学结合能为284.7、530.8、714.8和933.0 eV处分别对应的是C1s、O1s、Fe2p和Cu2p轨道, 说明材料中包含这4种元素.在图 9(a)的插表中展示了反应前后Cu0@ Fe3 O4中4种元素的原子百分比, 可以看出反应后铜元素的占比降低, 可能是由于少量Fe3 O4壳剥离后, Cu0暴露后被腐蚀.在图 9(b)中, Cu2p的XPS光谱在953.87 eV和933.97 eV处的特征峰分别归因于Cu0的Cu2p1/2和Cu2p3/2的自旋轨道.图 9(c)为Fe 2p的高分辨率XPS光谱图, 结合能为712.46 eV和710.53 eV处被分别认定为Fe3+和Fe2+.反应前后Fe3+和Fe2+的峰面积对比可看出, Fe2+的占比增加了微弱的0.6%.在铁基催化剂的催化反应中, Fe(Ⅲ)/Fe(Ⅱ)的氧化还原电势(0.77 V)比SO5-·/HSO4-的还原电势(1.1 V)低, 使得Fe(Ⅱ)的还原在热力学上不可行[式(13)], 通常是反应的限制条件.Fe3+/Fe2+的氧化还原电位高于Cu2+/Cu+[式(14)和(15)], 因此Fe3+在Cu+的作用下还原成Fe2+, 在热力学上是可行的, 反应后催化剂XPS光谱中Fe2+占比微弱增强的现象证实了Fe3+还原的存在[式(16)].因此, 铜的存在能够促进Fe2+的还原, 从而提高体系的反应持久性和稳定性.
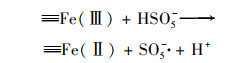
|
(13) |
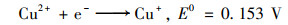
|
(14) |
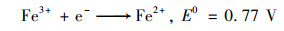
|
(15) |
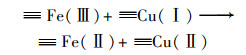
|
(16) |

|
图 9 Cu0@ Fe3 O4的XPS全扫描图以及在Cu 2p、Fe 2p和O 1s的高分辨率XPS光谱 Fig. 9 Full spectrum and high resolution XPS spectra of Cu 2p, Fe 2p, and O 1s |
另外, 在图 9(d)中, 反应前后催化剂O 1s的XPS光谱图被测定.结合能在530.07 eV附近的主峰归因于晶格氧(M—O), 在反应前的Cu0@ Fe3 O4中, M—O占所有表面氧的55%.反应后, M—O的占比降低至45%, 而531.4 eV附近的羟基氧(O—H)和532.2 eV附近的吸附氧(H2 O)占比均增加, 表明Cu0@ Fe3 O4在降解实验中被强烈羟基化.根据Sui等的研究, 表面羟基是催化分解和产生活性自由基的活性位点[31].因此大量的表面羟基有利于增强反应的氧化降解.
2.4.3 非均相活化机制根据以上研究结果提出了可能的Cu0@ Fe3 O4活化PMS机制(图 10), 该机制主要涉及两个方面.一方面, 零价铜能够被氧化为Cu(Ⅰ)[式(17)], 然后Cu(Ⅰ)和Fe(Ⅱ)可以提供电子活化PMS生成SO4-·和HO·[式(18)~(21)].在一定程度上, 高价铜和铁能够被PMS还原为低价态的Cu(Ⅰ)和Fe(Ⅱ)[式(22)和式(23)], 从而使Cu0@ Fe3 O4能够持续被催化.与此同时, 体系中的SO4-·可以和H2 O/OH-反应生成HO·[式(24)和式(25)].另一方面, 被证实的铁铜双金属协同作用下, 部分Fe(Ⅲ)能被Cu(Ⅰ)还原为Fe(Ⅱ), 从而使Cu0@ Fe3 O4/PMS体系形成完整的氧化还原循环[式(16)].最终体系中生成的SO4-·和HO·攻击污染物PNP, 从而将其分解为不同的中间产物, 最终矿化为CO2和H2 O[式(26)].

|
(17) |
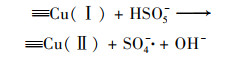
|
(18) |

|
(19) |
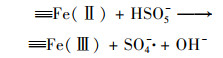
|
(20) |
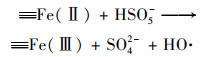
|
(21) |
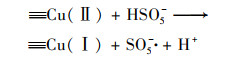
|
(22) |
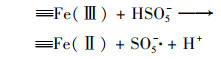
|
(23) |
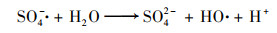
|
(24) |

|
(25) |
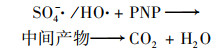
|
(26) |

|
图 10 Cu0@ Fe3 O4/PMS体系降解对-硝基苯酚的降解反应机制 Fig. 10 Reaction mechanism of Cu0@ Fe3 O4/PMS system for degradation of p-nitrophenol |
Cu0@ Fe3 O4/PMS体系反应是非均相过程, 与Cu0/PMS体系依赖腐蚀浸出铜离子激活PMS从而降解污染物的作用机制不同, 因此不依赖酸性pH环境, 具有宽泛的pH适应范围.
3 结论(1) 在自然pH值(5.65)下, 200 mg·L-1 Cu0@ Fe3 O4和0.5 mmol·L-1 PMS在60 min内对5 mg·L-1 PNP的降解率可达到96%.
(2) Cu0@ Fe3 O4的核壳结构改变了单纯Cu0在PMS存在时依赖酸性pH腐蚀释放出铜离子从而生成活性自由基的机制, 极大程度地降低了铜的浸出, 具有宽泛的pH适应范围.Cu0@ Fe3 O4/PMS体系降解污染物是非均相界面反应.
(3) Cu0@ Fe3 O4/PMS体系中存在SO4-·和HO·两种活性自由基, 对反应体系的贡献率分别为34%和60%, HO·是主导自由基.
(4) 反应体系中Fe和Cu之间存在双金属协同催化作用, Cu(Ⅰ)的存在能够促进Fe(Ⅲ)还原为Fe(Ⅱ), 从而形成良好的氧化还原循环, 提高反应体系的持久性.
| [1] | Zhang H, Ji Q Q, Lai L D, et al. Degradation of p-nitrophenol (PNP) in aqueous solution by mFe/Cu-air-PS system[J]. Chinese Chemical Letters, 2019, 30(5): 1129-1132. DOI:10.1016/j.cclet.2019.01.025 |
| [2] |
代志峰, 赵同谦, 阴永光, 等. 硝酸纤维素膜光降解水中对硝基苯酚的机制[J]. 环境科学, 2019, 40(2): 685-692. Dai Z F, Zhao T G, Yin Y G, et al. Photolysis mechanism of p-nitrophenol by nitrocellulose membrane in aqueous solution[J]. Environmental Science, 2019, 40(2): 685-692. |
| [3] | Chun C L, Baer D R, Matson D W, et al. Characterization and reactivity of iron nanoparticles prepared with added Cu, Pd, and Ni[J]. Environmental Science & Technology, 2010, 44(13): 5079-5085. |
| [4] | Gong J Y, Lee C S, Chang Y Y, et al. Novel self-assembled bimetallic structure of Bi/Fe0:the oxidative and reductive degradation of hexahydro-1, 3, 5-trinitro-1, 3, 5-triazine (RDX)[J]. Journal of Hazardous Materials, 2015, 286: 107-117. DOI:10.1016/j.jhazmat.2014.10.063 |
| [5] |
李晶, 鲍建国, 杜江坤, 等. Fe/Cu双金属活化过一硫酸盐降解四环素的机制[J]. 环境科学, 2018, 39(7): 3203-3211. Li J, Bao J G, Du J K, et al. Degradation mechanism of tetracycline using Fe/Cu oxides as heterogeneous activators of peroxymonosulfate[J]. Environmental Science, 2018, 39(7): 3203-3211. |
| [6] |
徐劼, 王琳, 陈家斌, 等. 磁性Fe3O4-CuO非均相活化过碳酸钠降解AO7[J]. 环境科学, 2020, 41(4): 1734-1742. Xu J, Wang L, Chen J B, et al. Degradation of AO7 with magnetic Fe3O4-CuO heterogeneous catalyzed sodium percarbonate system[J]. Environmental Science, 2020, 41(4): 1734-1742. |
| [7] | Goyal A, Sharma R, Bansal S, et al. Functionalized core-shell nanostructures with inherent magnetic character:Outperforming candidates for the activation of PMS[J]. Advanced Powder Technology, 2018, 29(2): 245-256. DOI:10.1016/j.apt.2017.11.008 |
| [8] | Mitsudome T, Kaneda K. Advanced core-shell nanoparticle catalysts for efficient organic transformations[J]. ChemCatChem, 2013, 5(7): 1681-1691. DOI:10.1002/cctc.201200724 |
| [9] | Yan W L, Herzing A A, Kiely C J, et al. Nanoscale zero-valent iron (nZVI):aspects of the core-shell structure and reactions with inorganic species in water[J]. Journal of Contaminant Hydrology, 2010, 118(3-4): 96-104. DOI:10.1016/j.jconhyd.2010.09.003 |
| [10] | Wang Y, Ke Y C, Li J Z, et al. Dispersion behavior of core-shell silica-polymer nanoparticles[J]. China Particuology, 2007, 5(4): 300-304. DOI:10.1016/j.cpart.2007.04.004 |
| [11] | Zhang M Y, Sheng Q L, Nie F, et al. Synthesis of Cu nanoparticles-loaded Fe3O4@carbon core-shell nanocomposite and its application for electrochemical sensing of hydrogen peroxide[J]. Journal of Electroanalytical Chemistry, 2014, 730: 10-15. DOI:10.1016/j.jelechem.2014.07.020 |
| [12] | Zhu M Y, Diao G W. Synthesis of porous Fe3O4 nanospheres and its application for the catalytic degradation of xylenol orange[J]. The Journal of Physical Chemistry C, 2011, 115(39): 18923-18934. DOI:10.1021/jp200418j |
| [13] | Keyhanian F, Shariati S, Faraji M, et al. Magnetite nanoparticles with surface modification for removal of methyl violet from aqueous solutions[J]. Arabian Journal of Chemistry, 2016, 9(Suppl 1): S348-S354. |
| [14] | Zhou P, Zhang J, Liu J L, et al. Degradation of organic contaminants by activated persulfate using zero valent copper in acidic aqueous conditions[J]. RSC Advances, 2016, 6(101): 99532-99539. DOI:10.1039/C6RA24431A |
| [15] | Zhou P, Zhang J, Zhang Y L, et al. Degradation of 2, 4-dichlorophenol by activating persulfate and peroxomonosulfate using micron or nanoscale zero-valent copper[J]. Journal of Hazardous Materials, 2018, 344: 1209-1219. DOI:10.1016/j.jhazmat.2017.11.023 |
| [16] | Nie G, Huang J, Hu Y Z, et al. Heterogeneous catalytic activation of peroxymonosulfate for efficient degradation of organic pollutants by magnetic Cu0/Fe3O4 submicron composites[J]. Chinese Journal of Catalysis, 2017, 38(2): 227-239. |
| [17] | Pham V L, Kim D G, Ko S O. Oxidative degradation of the antibiotic oxytetracycline by Cu@Fe3O4 core-shell nanoparticles[J]. Science of the Total Environment, 2018, 631-632: 608-618. DOI:10.1016/j.scitotenv.2018.03.067 |
| [18] | Pham V L, Kim D G, Ko S O. Cu@Fe3O4 core-shell nanoparticle-catalyzed oxidative degradation of the antibiotic oxytetracycline in pre-treated landfill leachate[J]. Chemosphere, 2018, 191: 639-650. DOI:10.1016/j.chemosphere.2017.10.090 |
| [19] | Zhang S W, Fan Q H, Gao H H, et al. Formation of Fe3O4@MnO2 ball-in-ball hollow spheres as a high performance catalyst with enhanced catalytic performances[J]. Journal of Materials Chemistry A, 2016, 4(4): 1414-1422. DOI:10.1039/C5TA08400H |
| [20] | Ghauch A, Ayoub G, Naim S. Degradation of sulfamethoxazole by persulfate assisted micrometric Fe0 in aqueous solution[J]. Chemical Engineering Journal, 2013, 228: 1168-1181. |
| [21] | Alder M G, Hill G R. The kinetics and mechanism of hydroxide ion catalyzed ozone decomposition in aqueous solution[J]. Journal of the American Chemical Society, 1950, 72(5): 1884-1886. DOI:10.1021/ja01161a007 |
| [22] | Huang K C, Couttenye R A, Hoag G E. Kinetics of heat-assisted persulfate oxidation of methyl tert-butyl ether (MTBE)[J]. Soil and Sediment Contamination:An International Journal, 2002, 11(3): 447-448. DOI:10.1080/20025891107717 |
| [23] | Anipsitakis G P, Dionysiou D D, Gonzalez M A. Cobalt-mediated activation of peroxymonosulfate and sulfate radical attack on phenolic compounds. implications of chloride ions[J]. Environmental Science & Technology, 2006, 40(3): 1000-1007. |
| [24] | Lei X, Xu C, Zhao M R, et al. Oxidative removal of aqueous steroid estrogens by manganese oxides[J]. Water Research, 2008, 42(20): 5038-5044. DOI:10.1016/j.watres.2008.09.016 |
| [25] | Lin K D, Liu W P, Gan J. Oxidative removal of bisphenol a by manganese dioxide:efficacy, products, and pathways[J]. Environmental Science & Technology, 2009, 43(10): 3860-3864. |
| [26] | Kolthoff I M, Miller I K. The chemistry of persulfate. Ⅰ. The kinetics and mechanism of the decomposition of the persulfate Ion in aqueous medium[J]. Journal of the American Chemical Society, 1951, 73(7): 3055-3059. DOI:10.1021/ja01151a024 |
| [27] | Zhou D N, Chen L, Li J J, et al. Transition metal catalyzed sulfite auto-oxidation systems for oxidative decontamination in waters:a state-of-the-art minireview[J]. Chemical Engineering Journal, 2018, 346: 726-738. DOI:10.1016/j.cej.2018.04.016 |
| [28] | Zhou L, Zheng W, Ji Y F, et al. Ferrous-activated persulfate oxidation of arsenic(Ⅲ) and diuron in aquatic system[J]. Journal of Hazardous Materials, 2013, 263: 422-430. DOI:10.1016/j.jhazmat.2013.09.056 |
| [29] | Du J K, Bao J G, Liu Y, et al. Efficient activation of peroxymonosulfate by magnetic Mn-MGO for degradation of bisphenol A[J]. Journal of Hazardous Materials, 2016, 320: 150-159. DOI:10.1016/j.jhazmat.2016.08.021 |
| [30] |
苏跃涵, 张利朋, 王枫亮, 等. Fe2+/单过氧硫酸氢盐(PMS)体系降解水体中酮洛芬的机制研究[J]. 环境化学, 2016, 35(9): 1753-1761. Su Y H, Zhang L P, Wang F L, et al. Degradation mechanism of ketoprofen by Fe2+/potassium peroxy monosulfate (PMS) oxidation process in aqueous[J]. Environmental Chemistry, 2016, 35(9): 1753-1761. |
| [31] | Sui M H, Sheng L, Lu K X, et al. FeOOH catalytic ozonation of oxalic acid and the effect of phosphate binding on its catalytic activity[J]. Applied Catalysis B:Environmental, 2010, 96(1-2): 94-100. DOI:10.1016/j.apcatb.2010.02.005 |