 2020, Vol. 41
2020, Vol. 41



2. 上海市污染防治与生态安全研究院, 上海 200092
2. Shanghai Institute of Pollution Control and Ecological Safety, Shanghai 200092, China
萘系磺酸化合物是指以萘环为母核, 含有一个或多个磺酸基团(—SO3H)的化合物, 芳环上还可能含有其他取代基如氨基和羟基等[1].这类有机化合物在工业上应用广泛, 尤其用于合成直接、活性染料, 但同时导致了15%的染料转移至废水中[2].从结构上看, 磺酸基团的存在使这类化合物在水中具有高度流动性, 同时萘环和碳硫键的稳定结构导致了顽固性, 这意味着萘磺酸化合物能够在环境中累积并最终可能导致生态毒理学风险.
用物理法处理废水方便快捷, 非常灵活.实际应用中常用的物理手段是活性炭[3].多孔碳材料能从水溶液中快速地吸附萘[4], 但对含萘磺酸的废水, 有研究发现活性炭柱对H酸的吸附容量较低[5].这是由于活性炭表层含有大量碱性基团, 与吸电子的磺酸基团相排斥, 降低了吸附亲和力, 并且排斥作用随着磺酸基团的增加而增强, 因此活性炭不适于吸附萘磺酸.Jia等[6]用碱性树脂吸附去除水中的2-萘磺酸, 也发现磺酸基团的存在可能不利于树脂的吸附作用.石墨烯[7]以及合成的聚丙烯酯聚合物[8]等也能用于萘磺酸的去除.为了资源回收, 综合萃取法也较为常见[1].物理手段的有效性毋庸置疑, 但吸附剂相对昂贵、再生耗能较大以及潜在的二次污染问题是其缺陷[9].生物法处理萘磺酸废水的研究较少, 早期的文献已经指出萘磺酸几乎不能被微生物降解[10].这不仅表现在极端的酸性以及高盐条件, 更重要的是萘磺酸进入细胞后通过氮化碱基的取代作用造成DNA损伤, 抑制了微生物的生长繁殖[11].同时也说明环境介质中累积的萘磺酸无法依靠自然条件进行降解[12].化学法特殊优势在于反应速度快, 去除效率高, 反应彻底.臭氧法[13]和芬顿法[14]已经被用于氧化降解萘磺酸, 而且COD均得到有效去除, 可生化性显著升高, 因此氧化降解值得研究.
过硫酸盐(PS, E0=2.01 V)是当前环境领域研究的热点.迄今为止PS的研究大多偏向于新材料开发, 很少考虑实际应用性[15].据笔者所知, 目前还没有研究用PS氧化降解萘磺酸的文献.相对传统芬顿, 虽然PS的反应性略差, 但PS不仅价格便宜, 性质稳定利于储存运输, 而且可以在较宽的pH范围内使用, 最关键的是可灵活使用不同方式活化PS获得氧化性更强的硫酸根自由基(SO4- ·, E0=2.6 V), 其半衰期较长, 反应速率可控[15, 16].笔者之前的研究表明, 碱性条件下PS氧化降解H酸的效果更好, 这与其他研究一致[17], 归因于碱性条件下羟基自由基(·OH, E0=2.7 V)大量产生[18].相比NaOH溶液, CaO用作活化剂的原因一方面在于价格便宜, 萘磺酸废水的酸性条件利于其溶解, 产生的热量也能加速氧化, 另一方面Ca2+的存在可产生部分磺酸钙、硫酸钙降低生物毒性, 为后续生化创造了可能性.本文以H酸为特征污染物, 对比了碱活化、热活化以及碱热复合活化PS对H酸的降解效果, 并探讨了复合活化中的影响因素及H酸降解机制, 旨在为活化PS技术用于含萘磺酸废水的实际处理提供参考.
1 材料与方法 1.1 实验试剂H酸一钠盐, 即1-氨基-8-萘酚-3, 6-二磺酸单钠盐(≥95%)购于Shanghai yuanye Bio-Technology公司, 分子结构见图 1; 过硫酸钠(≥98%)购于sinopharm chemical reagent公司; 氧化钙(≥98%)购于Macklin公司; 甲醇(色谱纯)购于Aladdin公司, 氢氧化钠, 硫酸, 氯化钠, 硫酸钠均为分析纯, 实验用水为去离子水.

|
图 1 H酸分子结构示意 Fig. 1 H acid molecular structure |
氧化反应均在250 mL锥形瓶中进行, H酸的初始质量浓度为500 mg ·L-1, 150 mL.溶液pH通过1 mol ·L-1的H2SO4或NaOH进行调节.氧化钙加入锥形瓶后使用磁力搅拌器搅拌数分钟, 溶解后放入摇床中, 转速为150 r ·min-1, 设置不同的温度并预热数分钟后再加入PS进行反应.分别在0、5、10、20、40、60、80和100 min取2.5 mL反应液过滤后与0.1 mL甲醇混合, 再将样品置于冰水浴中淬灭, 用分光光度计分析样品中的H酸质量浓度.H酸质量浓度0~40 mg ·L-1范围内线性关系良好, 该范围内回归方程为: y=0.019x+0.032, R2=0.996, 见图 2.

|
图 2 H酸在358 nm处吸收光谱标准曲线 Fig. 2 Standard curve of absorption spectrum of H acid at 358 nm |
在上述体系中分别考察了氧化钙质量浓度(500、750、1 000和1 250 mg ·L-1), 温度(35、45、55和65℃), PS浓度(3、6、9和12 mmol ·L-1), 无机阴离子(Cl-、SO42-和CO32-), CaCl2质量浓度(1 000、1 500、2 000和2 500 mg ·L-1)以及溶液初始pH(3、5、7、9和11)对H酸降解影响.测定H酸溶液及实际废水中的TOC时, 样品在过滤后仅采用冰水浴以终止反应.进行了动力学的研究, 用式(1)对H酸的降解进行伪一级拟合.
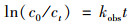
|
(1) |
式中, c0与ct分别是H酸在溶液中的初始质量浓度及t时刻的残留质量浓度(mg ·L-1); kobs为伪一级反应速率常数(min-1); t为反应时间(min).
在热活化体系中还通过Arrhenius方程研究计算了H酸表观活化能, 见式(2):
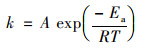
|
(2) |
式中, A为指前因子(min-1), Ea为反应的表观活化能(kJ ·mol-1), R为气体常数[8.314 J ·(mol ·K)-1], T为绝对温度(K).
1.3 分析方法溶液中的H酸残留质量浓度经过滤后采用分光光度计测定, 分析波长为358 nm[14].耶拿multi N/C 3100 TOC仪测定反应液中残余的TOC质量浓度.GC-MS(Agilent 7890A-5975C, USA)对H酸的降解中间产物进行分析鉴定, 配置HP-5MS(Agilent 19091S-433, 30 m×0.25 mm×0.25 μm)色谱柱, 进样口温度为250℃, 氦气载气, 流量为1.00 mL ·min-1, 柱箱初始温度为80℃, 保持4 min, 以2℃ ·min-1升至200℃, 保持20 min, 再以5℃ ·min-1升至280℃, 保持6 min.
2 结果与讨论 2.1 碱活化在H酸质量浓度=500 mg ·L-1, T=25℃, PS=6 mmol ·L-1条件下, 考察了CaO投加量对H酸降解的影响.如图 3(a)所示, 空白条件下, H酸去除率仅有42.5%, 随着CaO投加量从500增加至1250 mg ·L-1时, H酸降解率也由69.4%提升至82.8%, 反应后溶液pH维持在12左右, 见图 3(b).H酸的去除率提升可能与溶液pH改变有关.

|
图 3 CaO投加量对H酸降解及溶液pH影响 Fig. 3 Effect of CaO dosage on H acid degradation and pH of solution |
随着CaO的溶解, 反应体系迅速由酸性变为强碱性.PS在碱性条件下水解为SO52-和SO4- ·, 不稳定的SO52-裂解产生的HO2-与PS反应使得溶液中SO4- ·以及O2- ·迅速增加, 并通过电子传递作用生成氧化性更强的·OH, 见式(3)~(6), 最终促进了污染物的降解[19, 20].
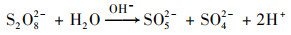
|
(3) |
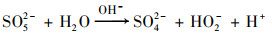
|
(4) |
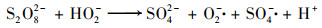
|
(5) |

|
(6) |
在H酸降解过程中, CaO既能调节反应液pH又能产生钙盐沉淀, 均可能影响H酸的降解, 因此研究了初始pH以及Ca2+对降解的影响, 图 4显示当初始pH由酸性增加至中性时, H酸的降解被抑制, 在pH为7时抑制程度最大, 但随着溶液pH值继续增加, 反而促进了H酸的降解.这可能是因为pH的改变影响了自由基的种类及产生速率[21]. Liang等[22]的研究证明, 当pH < 7时, SO4- ·占主导地位, 同时·OH随pH增大而增加, 当pH>12时, ·OH占主导.这解释了PS降解有机污染物与溶液pH之间的依赖性, 也能用于解释H酸降解与pH之间的联系.磺酸钙和硫酸钙可能吸附H酸, 为了研究这一作用, 往反应液中加入一定量的CaCl2探讨Ca2+对H酸的降解影响, 反应条件为H酸质量浓度为500 mg ·L-1, 温度为25℃, PS浓度为6 mmol ·L-1, CaCl2质量浓度为1 000~2 500 mg ·L-1, 但图(5)的结果说明钙盐沉淀的影响有限.

|
图 4 初始pH对H酸降解影响 Fig. 4 Effect of initial pH on H acid degradation |

|
图 5 Ca2+对H酸降解影响 Fig. 5 Effect of Ca2+ on H acid degradation |
温度对活化PS产生SO4- ·和·OH具有重要的影响.研究了不同温度对H酸的降解影响, 如图 6(a).结果表明, 当PS浓度为6 mmol ·L-1, 反应100 min时, 观察到25、35、45、55和65℃对应的H酸去除率依次为42.5%、51.4%、63.3%、73.5%和77.5%, 图 6(b)中反应速率常数分别为4.92×10-3(R2=0.855)、6.99×10-3(R2=0.91)、8.97×10-3(R2=0.855)、1.46×10-2(R2=0.818)和2.20×10-2(R2=0.884).H酸的去除率与温度正相关, 这与其他报道一致[23, 24].由于加热使O—O间成键电子对均裂, 理论上1 mol PS可产生2 mol的SO4- ·, 见式(7), 并且随着温度升高, 分解速率加快, SO4- ·和·OH的产生速率也加快, 最终提高污染物的降解速率[18]. Johnson等[25]评估了不同温度下PS的持久性, 认为温度上升(>50℃)导致PS分解速率明显加快, 但不利于PS在环境介质中的扩散.从降解污染物和经济性角度考虑, 合适的活化温度应在50~60℃.

|
图 6 温度对H酸降解影响 Fig. 6 Effect of temperature on H acid degradation |
通过阿伦尼乌斯模型, 图 6(c), 计算出降解H酸的表观活化能为37.85 kJ ·mol-1, 温度升高对其降解有很大的促进作用.

|
(7) |
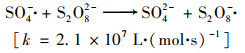
|
(8) |
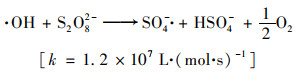
|
(9) |
上述实验证明碱和热均能活化PS并对H酸的氧化降解有着良好的效果. Huang等[26]的研究发现碱热协同可以快速实现污染物的降解.为了进一步提高H酸去除效果, 对碱热复合活化进行探讨.在CaO投加量为500 mg ·L-1, PS浓度为6 mmol ·L-1条件下研究了不同温度(25、45和65℃)下H酸的降解行为.图 7显示, 在CaO质量浓度一定时, 温度升高40℃ H酸去除率仅提高3.1%, 说明在复合活化中温度可能影响较小.图 8进一步对比发现, 当复合活化中温度低于50℃时对H酸去除效果优于热活化, 但随着温度高于50℃时复合活化对H酸的去除效果不如热活化.复合活化中SO4- ·来源于碱水解以及热活化, 同时碱活化中会产生其他活性物种如·OH和O2- ·.热活化中由于反应液呈酸性, 可认为活性物种仅为SO4- ·.同温度下, 复合活化产生的活性物种浓度应高于热活化, 但复合活化在高温下对H酸的去除效果不如热活化.这可能是碱热共同作用下PS分解过快, 加剧了副反应[27].可以考虑改变PS的浓度.

|
图 7 碱热复合活化对H酸降解效果 Fig. 7 Effect of alkali thermal complex activation on H acid degradation |

|
图 8 碱热复合活化与热活化对比 Fig. 8 Comparison of alkali thermal complex activation and thermal activation |
PS浓度是复合活化中重要的影响因素, 在CaO投加量为500 mg ·L-1, 反应温度为45℃条件下, 如图 9, PS初始浓度为3、6、9和12 mmol ·L-1反应100 min时H酸的去除率依次为68.7%、69.4%、71.5%以及73.8%.在前10 min, 高浓度下PS对H酸的降解速率更快, 证明该条件下高浓度的PS能够迅速分解, 但并未大幅提高H酸的降解效果, 这可能是中间产物或副产物等非目标污染物的影响[28].

|
图 9 PS浓度对H酸降解影响 Fig. 9 Effect of PS concentration on the degradation of H acid |
实际废水成分复杂, 其中一些有机或无机化合物可作为SO4- ·和·OH的清除剂[29].复合活化PS体系中, 在CaO投加量为500 mg ·L-1, 反应温度为45℃, PS初始浓度为6 mmol ·L-1条件下研究了浓度均为60 mmol ·L-1的Cl-、CO32-和SO42-对H酸降解行为的影响, 结果见图 10.从中可知, CO32-存在下, 反应100 min时H酸去除率从69.4%降至62.1%.在活化PS体系中, 由于SO4- ·和·OH与CO32-均具有较大的反应速率, 能被CO32-消耗, 因此影响了H酸的降解, 见式(10)~(11)[18]. Wang等[30]的研究发现在Fe2+/PS体系中, Cl-的存在对醋氨酚的降解表现出双重作用, 高浓度Cl-(10 mmol ·L-1)促进醋氨酚的降解, 此时体系中主要的活性物种是Cl2- ·而不是SO4- ·.有研究也报道了Cl-的双重作用[31, 32].但本研究中没有观察到Cl-抑制H酸降解.H酸上含有亲水性的磺酸基团, 在反应溶液中易脱落, 为了研究活化PS体系中SO42-对H酸降解行为影响, 往碱热体系中加入60 mmol ·L-1 SO42-, 结果发现SO42-不影响H酸的降解.

|
图 10 无机阴离子对H酸降解影响 Fig. 10 Effect of inorganic anions on the degradation of H acid |
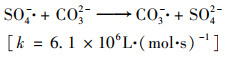
|
(10) |
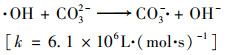
|
(11) |
利用复合活化PS探究H酸的矿化情况.图 11说明随着时间推移, 矿化度逐渐提高, 反应100 min时, TOC去除率达到16%, 此时体系中CaO投加量为500 mg ·L-1, 反应温度为45℃, PS浓度为6.0 mmol ·L-1.相比H酸去除率, 矿化率并不理想.这可能是由于主要的反应发生在侧链, 并未实现苯环开环[33]. Zhu等[34]用芬顿法(H2 O2 :Fe2+=20 :1)氧化降解染料中间体1-偶氮-2-萘酚-4-磺酸(1 mmol ·L-1), 发现即使H2 O2浓度高达30 mmol ·L-1处理200 min, TOC仅仅从110 mg ·L-1降至82 mg ·L-1, 矿化率为25.4%.相比之下, H2 O2投加量是PS的5倍, 但对萘磺酸的矿化率仅增加了9.4%.

|
图 11 降解过程中H酸的矿化度 Fig. 11 Mineralization degree of H acid during degradation |
碱热复合活化体系下, 分别在0、2、10、30、60和100 min时对H酸反应液进行扫描.图 12中0 min吸收曲线在200~250 nm处出现末端吸收, 吸收峰1对应的吸收波长λ为358 nm, 这是H酸结构中的萘环[14].PS加入仅2 min后该特征峰即消失, 表明H酸中的萘环迅速裂解, 同时可见光400~700 nm范围内的吸收首先大幅增加, 随着反应进行又逐渐减弱.溶液颜色由浅黄色变为血红色, 最终变为黑红色, 这种现象表明降解过程中产生了生色团.这与Liu等[14]和Zhu等[35]的研究结果一致.

|
图 12 H酸降解中间产物UV-vis光谱扫描 Fig. 12 UV-vis spectral scan of H acid degradation intermediates |
取t为5、30和100 min的降解样品, 利用GC-MS对复合活化PS降解H酸的中间产物进行初步鉴定发现了较多的中间体.由于缺乏挥发性, 磺酸基团可能难以通过气相检测, 但是将产物硅烷化后仍鉴定出碳酸、1, 4-苯二甲酸和3, 4-二甲基苯甲酸等产物, 相应的保留时间分别为8.114、47.059和104.331 min, 见图 13.萘环化学性质比苯活泼, 其α位的电子云密度较大, 容易发生羟基化.同理, H酸中富含电子及大量不饱和键, 易被SO4- ·及·OH通过不同的形式破坏, 例如直接攻击烯烃双键形成羧酸衍生物等[14], 或萘醌化[36], 但本研究中由于萘磺酸

|
图 13 H酸降解产物硅烷化后总离子色谱图 Fig. 13 Total ion chromatogram of alkylated H acid degradation product |
较弱的挥发性以及样品处理过程的不确定性并未找到萘醌.与Liu等[14]的研究不同的是, 本研究在产物中发现了对-苯二甲酸. Zhu等[34]在芬顿氧化萘磺酸研究中也发现了邻-苯二甲酸酐的存在, 这最可能来源于邻-苯二甲酸酐, 相比萘醌, 前者氧化更为彻底, 这可能也说明PS的氧化体系相比芬顿更彻底.H酸降解路径见图 14, H酸在SO4- ·和·OH攻击下迅速被氧化为中间产物邻-苯二甲酸酐, 同时由于水解和异构等作用转化为更为稳定的1, 4-苯二甲酸, 这可能是矿化度不高的原因.

|
图 14 H酸可能的降解途径 Fig. 14 Possible degradation pathway of H acid |
(1) 氧化钙可以用于活化PS处理萘系磺酸盐废水, 投加量与H酸的去除率正相关.碱活化H酸去除效率、反应速率均优于热活化.
(2) 碱热复合活化大幅提高了反应速率, 但PS迅速分解导致副反应增加, 即使提高PS浓度对H酸去除率提升仍不明显.无机离子中CO32-离子的存在抑制了H酸降解, 同浓度下Cl-和SO42-的影响不大.
(3) H酸降解过程中颜色变化明显, 产生了大量的生色团, 中间产物中发现了较稳定的对-苯二甲酸.
| [1] |
孙培志.萘磺酸废水的处理研究[D].北京: 北京化工大学, 2018. Sun P Z. Study on treatment of naphthalene sulfonic acid wastewater[D]. Beijing: Beijing University of Chemical Technology, 2018. |
| [2] | Khataee A R, Kasiri M B. Photocatalytic degradation of organic dyes in the presence of nanostructured titanium dioxide:influence of the chemical structure of dyes[J]. Journal of Molecular Catalysis A:Chemical, 2010, 328(1-2): 8-26. |
| [3] | Rocha L S, Pereira D, Sousa é, et al. Recent advances on the development and application of magnetic activated carbon and char for the removal of pharmaceutical compounds from waters:a review[J]. Science of the Total Environment, 2020, 718: 137272. |
| [4] | Yuan J, Feng L, Wang J X. Rapid adsorption of naphthalene from aqueous solution by naphthylmethyl derived porous carbon materials[J]. Journal of Molecular Liquids, 2020, 304: 112768. |
| [5] | Rivera-utrilla J, Sánchez-polo M. Degradation and removal of naphthalenesulphonic acids by means of adsorption and ozonation catalyzed by activated carbon in water[J]. Water Resources Research, 2003, 39(9): 1232. |
| [6] | Jia D M, Li C H, Zhao B L, et al. Studies on the adsorption of 2-naphthalenesulfonic acid on basic resin from effluents[J]. Journal of Chemical & Engineering Data, 2010, 55(12): 5801-5806. |
| [7] | Wu T, Cai X, Tan S Z, et al. Adsorption characteristics of acrylonitrile, p-toluenesulfonic acid, 1-naphthalenesulfonic acid and methyl blue on graphene in aqueous solutions[J]. Chemical Engineering Journal, 2011, 173(1): 144-149. |
| [8] | Pan B J, Zhang W M, Pan B C, et al. Efficient removal of aromatic sulfonates from wastewater by a recyclable polymer:2-naphthalene sulfonate as a representative pollutant[J]. Environmental Science & Technology, 2008, 42(19): 7411-7416. |
| [9] | Li F, Chen J J, Hu X, et al. Applications of carbonaceous adsorbents in the remediation of polycyclic aromatic hydrocarbon-contaminated sediments:a review[J]. Journal of Cleaner Production, 2020, 255: 120263. |
| [10] | Kneifel H, Elmendorff K, Hegewald E, et al. Biotransformation of 1-naphthalenesulfonic acid by the green alga scenedesmus obliquus[J]. Archives of Microbiology, 1997, 167(1): 32-37. |
| [11] | Rivera-utrilla J, Sánchez-polo M, Mondaca M A, et al. Effect of ozone and ozone/activated carbon treatments on genotoxic activity of naphthalenesulfonic acids[J]. Journal of Chemical Technology & Biotechnology, 2002, 77(8): 883-890. |
| [12] | Qin J X, Dai X G, Zhou Y, et al. Desalting and recovering naphthalenesulfonic acid from wastewater with concentrated bivalent salt by nanofiltration process[J]. Journal of Membrane Science, 2014, 468: 242-249. |
| [13] | Zhu S Y, Zheng X S, Li D T. Ozonation of naphthalene sulfonic acids in aqueous solutions. Part I:elimination of COD, TOC and increase of their biodegradability[J]. Water Research, 2002, 36(5): 1237-1243. |
| [14] | Liu H H, Chen Q Y, Yu Y, et al. Influence of fenton's reagent doses on the degradation and mineralization of H-acid[J]. Journal of Hazardous Materials, 2013, 263: 593-599. |
| [15] | Lee J, Von Gunten U, Kim J H. Persulfate-based advanced oxidation:critical assessment of opportunities and roadblocks[J]. Environmental Science & Technology, 2020, 54(6): 3064-3081. |
| [16] | Yang L, Xue J M, He L Y, et al. Review on ultrasound assisted persulfate degradation of organic contaminants in wastewater:influences, mechanisms and prospective[J]. Chemical Engineering Journal, 2019, 378: 122146. |
| [17] | Pan X X, Yan L Q, Qu R J, et al. Degradation of the UV-filter benzophenone-3 in aqueous solution using persulfate activated by heat, metal ions and light[J]. Chemosphere, 2018, 196: 95-104. |
| [18] |
章晋门, 陈泉源, 杨慧敏. 过硫酸盐活化方式与氧化降解有机物效能及其在污染场地修复中的应用[J]. 化工进展, 2020, 39(1): 1-13. Zhang J M, Chen Q Y, Yang H M. Influence of persulfate activation on organic pollutant degradation and application in remediation of contaminated land[J]. Chemical Industry and Engineering Progress, 2020, 39(1): 1-13. |
| [19] | Furman O S, Teel A L, Watts R J. Mechanism of base activation of persulfate[J]. Environmental Science & Technology, 2010, 44(16): 6423-6428. |
| [20] | Dominguez C M, Rodriguez V, Montero E, et al. Abatement of dichloromethane using persulfate activated by alkali:a kinetic study[J]. Separation and Purification Technology, 2020, 241: 116679. |
| [21] | Norzaee S, Taghavi M, Djahed B, et al. Degradation of penicillin G by heat activated persulfate in aqueous solution[J]. Journal of Environmental Management, 2018, 215: 316-323. |
| [22] | Liang C J, Su H W. Identification of sulfate and hydroxyl radicals in thermally activated persulfate[J]. Industrial & Engineering Chemistry Research, 2009, 48(11): 5558-5562. |
| [23] | Hu C Y, Hou Y Z, Lin Y L, et al. Investigation of iohexol degradation kinetics by using heat-activated persulfate[J]. Chemical Engineering Journal, 2020, 379: 122403. |
| [24] | Milh H, Schoenaers B, Stesmans A, et al. Degradation of sulfamethoxazole by heat-activated persulfate oxidation:elucidation of the degradation mechanism and influence of process parameters[J]. Chemical Engineering Journal, 2020, 379: 122234. |
| [25] | Johnson R L, Tratnyek P G, Johnson R O. Persulfate persistence under thermal activation conditions[J]. Environmental Science & Technology, 2008, 42(24): 9350-9356. |
| [26] | Huang Z H, Ji Z Y, Zhao Y Y, et al. Treatment of wastewater containing 2-methoxyphenol by persulfate with thermal and alkali synergistic activation:kinetics and mechanism[J]. Chemical Engineering Journal, 2020, 380: 122411. |
| [27] | Angkaew A, Sakulthaew C, Satapanajaru T, et al. UV-activated persulfate oxidation of 17β-estradiol:implications for discharge water remediation[J]. Journal of Environmental Chemical Engineering, 2019, 7(2): 102858. |
| [28] | Crincoli K R, Green C, Huling S G. Sulfate radical scavenging by mineral surfaces in persulfate-driven oxidation systems:reaction rate constants and implications[J]. Environmental Science & Technology, 2020, 54(3): 1955-1962. |
| [29] | Niu L J, Xian G, Long Z Q, et al. MnCeOx/diatomite catalyst for persulfate activation to degrade organic pollutants[J]. Journal of Environmental Sciences, 2020, 89: 206-217. |
| [30] | Wang S L, Wu J F, Lu X Q, et al. Removal of acetaminophen in the Fe2+/persulfate system:kinetic model and degradation pathways[J]. Chemical Engineering Journal, 2019, 358: 1091-1100. DOI:10.1016/j.cej.2018.09.145 |
| [31] | Lu X, Shao Y S, Gao N Y, et al. Investigation of clofibric acid removal by UV/persulfate and UV/chlorine processes:kinetics and formation of disinfection byproducts during subsequent chlor(am)ination[J]. Chemical Engineering Journal, 2018, 331: 364-371. |
| [32] | Wang Q, Lu X H, Cao Y, et al. Degradation of bisphenol S by heat activated persulfate:kinetics study, transformation pathways and influences of co-existing chemicals[J]. Chemical Engineering Journal, 2017, 328: 236-245. |
| [33] |
王晓晓, 王兆慧, 柳建设. 热活化过硫酸盐氧化降解水体中泛影酸钠的研究[J]. 环境科学学报, 2019, 39(5): 1519-1526. Wang X X, Wang Z H, Liu J S. Degradation of sodium diatrizoate by thermally activated persulfate oxidation process[J]. Acta Scientiae Circumstantiae, 2019, 39(5): 1519-1526. |
| [34] | Zhu N W, Gu L, Yuan H P, et al. Degradation pathway of the naphthalene azo dye intermediate 1-diazo-2-naphthol-4-sulfonic acid using fenton's reagent[J]. Water Research, 2012, 46(12): 3859-3867. |
| [35] | Zhu W P, Yang Z H, Wang L. Application of ferrous-hydrogen peroxide for the treatment of H-acid manufacturing process wastewater[J]. Water Research, 1996, 30(12): 2949-2954. |
| [36] | Sun L Y, Lu H, Zhou J T. Degradation of H-acid by combined photocatalysis and ozonation processes[J]. Dyes and Pigments, 2008, 76(3): 604-609. |