Community Structure and Predictive Functional Analysis of Surface Water Bacterioplankton in the Danjiangkou Reservoir
浮游细菌是水生生态系统的重要组成部分, 其可通过同化、异化等代谢过程来影响该生态系统中的物质循环、污染物释放等生物地球化学循环过程[1~3].丹江口水库作为南水北调中线工程核心水源区, 其水质状况直接关系到受水区居民的饮水安全.长期监测表明, 由于存在农业面源、村镇生活污水等污染, 造成丹江口库区总氮超标[4, 5].有研究表明, 微生物介导的氮代谢是地球氮循环的主要驱动力[6, 7].高通量测序技术能高通量得到特定的DNA片段, 全面地展示生物群落的组成结构[8].针对丹江口库区浮游细菌, 目前本课题组和其他研究者已采用高通量测序技术(high throughput sequencing)技术等方法开展了相关研究, 初步探究了其群落组成和影响因素[5, 9~11], 但目前针对其功能, 特别是氮循环过程中细菌群落组成和功能研究较少开展[12].同时, 之前关于的高通量测序分析主要关注于细菌群落结构(α和β多样性), 研究细菌群落组成、分布特征及其影响因素等, 对其功能研究开展较少[9, 13~17].基于高通量测序的PICRUSt(phylogenetic investigation of communities by reconstruction of unobserved states)功能预测分析相较于宏基因组研究, 更方便快捷、成本也更低, 同时预测效果可靠性较高[18], 目前该方法已在湖泊、海洋微生物研究中得到良好的应用[19~23].本课题组已成功采用PICRUSt软件预测丹江口库区表层沉积物[24]和库滨带[25]细菌功能, 但目前丹江口库区浮游细菌功能研究尚未开展.
目前, PICRUSt功能预测分析在丹江口库区浮游细菌功能研究中未见报道, 其与高通量测序技术结合分析, 将为探明库区浮游细菌群落组成和功能提供重要帮助.因此, 本研究选取丹江口库区库心和渠首2个生态点位, 采用16S rDNA MiSeq高通量测序技术研究表层浮游细菌群落组成, 结合PICRUSt分析预测浮游细菌功能, 重点分析氮循环相关基因表达情况, 通过微生物学角度分析丹江口库区不同位点氮的循环差异, 以期为丹江口水库水环境保护提供参考依据.
1 材料与方法
1.1 样品采集与理化指标测定
根据丹江口水库的地理位置特征和人类活动影响程度, 选取属于河南省南阳市的丹江库区库心(样品编号为K, 坐标为32°45′06.16″N和111°34′45.09″E)和渠首(样品编号为Q, 位于陶岔渠首大坝取水口上游, 坐标为32°40′02.76″N和111°37′47.12″E)[5, 9].参照文献[26]于2016年5月用柱状采水器采集表层(深度50 cm)水样2 L, 所有点位均取平行样3组(分别用K1、K2、K3和Q1、Q2、Q3表示库心和渠首样品3组平行样品). 600 mL水样用于提取细菌总DNA, 置于预先灭菌容器中存于冰盒, 运回实验室后进行检测分析, 剩余水样用于水质指标测定.参照文献[26]测定水样pH值、总氮(TN)、氨氮(NH4+-N)、总磷(TP)、化学需氧量(COD)、高锰酸盐指数、溶解氧(DO)、透明度(SD)以及叶绿素a含量(Chla)等理化指标.
1.2 样品总DNA提取
600 mL表层新鲜水样经20 μm筛绢预过滤, 以除去大型浮游植物和浮游动物.预过滤后的水样经0.22 μm无菌微孔滤膜(Pall Life sciences, Mumbai, India)过滤, 用以收集浮游细菌, 将滤膜剪碎置于50 mL无菌离心管中.按照Omega Water DNA Kit (Omega, USA)的说明, 提取水体中的样品总DNA.
1.3 高通量测序
采用细菌通用引物338F和806R对16S rRNA基因的V3~V4区进行PCR扩增, 扩增的反应条件为: 94℃, 5 min; 30×(94℃, 30 s; 54℃, 30 s; 72℃, 45 s); 72℃, 10 min.扩增体系(20 μL)为:5×FastPfu Buffer 4 μL、2.5 mmol ·L-1 dNTPs 2 μL、Forward Primer(5 μmol ·L-1)0.8 μL、Reverse Primer(5 μmol ·L-1) 0.8 μL、FastPfu Polymerase 0.4 μL、DNA模板10 ng[5, 16].采用上海美吉生物医药科技有限公司的MiSeq PE300测序仪(Illumina Inc, San Diego, CA, USA)进行序列测定.
1.4 数据分析
1.4.1 高通量数据分析
将Illumina MiSeq测序得到的下机数据(Raw Data)经处理后得到Tags序列, 与数据库(Gold database)进行比对, 检测嵌合体序列, 并最终去除其中的嵌合体序列, 得到最终的有效数据(Effective Tags).采用QIIME(quantitative insights into microbial ecology)进行生物信息学分析, 根据序列的相似度, 将序列归为多个OTU(operational taxonomic unit)[27].为了得到每个OTU对应的物种分类信息, 采用RDP classifier贝叶斯算法对97%相似水平的OTU代表序列进行分类学分析, 并在各个分类水平统计每个样品的群落组成.比对分类使用SILVA数据库[28].选取相似度在97%条件下的OTU生成预期的稀释曲线[29].利用UPGMA聚类分析进行细菌群落分布、聚类分析.采用STAMP软件[30]比较分析不同样品细菌群落结构的差异(P<0.05显著性差异和P<0.01极显著差异).
1.4.2 PICRUSt功能预测分析
功能和代谢途径预测采用PICRUSt软件进行分析, 利用QIIME获得的closed OTU-table与COG、KEGG数据库进行比对, 获得不同的数据库功能预测信息, 具体分析步骤基于在线分析平台(http://picrust.github.io/picrust/)[18].
2 结果与分析
2.1 丹江口库区水质理化性质
2016年5月对丹江口水库库心、渠首的理化监测结果表明各水体的水质总体较好, 除TN和高锰酸盐指数其他各项指标符合《地表水环境质量标准》(GB 38382-2002)Ⅰ类水标准要求(表 1).库心、渠首样品TN均超过0.90 mg ·L-1, 为Ⅲ类地表水标准.库心、渠首样品高锰酸盐指数均超过3.00 mg ·L-1, 为Ⅱ类地表水标准.长期监测表明, 丹江口水库水质总体较好, 符合《地表水环境质量标准》(GB 38382-2002)Ⅱ类(饮用水源)标准, 但存在农业面源、工业废水以及村镇生活污水等污染, 其中TN明显超标[4, 5], 本实验监测结果与上述结果一致.
表 1
(Table 1)

表 1 各样品主要理化参数
Table 1 Physical and chemical properties of the samples
| 样品 |
pH值 |
高锰酸盐指数/mg·L-1 |
TN /mg·L-1 |
NH4+-N /mg·L-1 |
TP /mg·L-1 |
COD /mg·L-1 |
Chla /mg·m3 |
SD /m |
DO /mg·L-1 |
| 库心 |
8.50±0.02 |
3.46±0.01 |
0.96±0.01 |
0.069±0.009 |
0.02 |
11.60±0.10 |
2.13±0.06 |
5 |
9.17±0.06 |
| 渠首 |
8.70±0.01 |
3.81±0.01 |
0.91±0.01 |
0.041±0.005 |
0.02 |
13.97±0.25 |
2.42±0.03 |
2.99±0.01 |
7.50±0.03 |
|
表 1 各样品主要理化参数
Table 1 Physical and chemical properties of the samples
|
2.2 不同样品浮游细菌群落结构分析
2.2.1 高通量测序结果及多样性的评估
高通量测序结果表明, 样品平均测序序列条数为25 582, 样品的文库覆盖率(good's coverage)高于99.50%, 在测序条数达到20 000条以上时稀释性曲线趋向平稳, 表明测序数据量合理, 能够代表物种的丰富度.采用群落丰富度指数Chao1和Ace、群落多样性指数Shannon指数、Simpson指数和文库覆盖率等对丹江口库区浮游细菌群落进行评估, 结果表明样品具有丰富的群落组成, 其群落多样性均较高(表 2).综合Sobs、丰富度指数Chao1和Ace以及多样性指数Simpson、Shannon、文库覆盖率, 渠首样品浮游细菌群落多样性高于库心样品.
表 2
(Table 2)
表 2 不同样品浮游细菌多样性评估表
Table 2 Estimation of rhizobacteria community diversity in different samples
| 样品 |
序列条带数 |
Sobs指数 |
Shannon指数 |
Simpson指数 |
Ace指数 |
Chao 1指数 |
覆盖率/% |
| 库心 |
27 098±4 940 |
355±29 |
4.13±0.09 |
0.027 9±0.002 7 |
426±23 |
437±37 |
99.70±0.05 |
| 渠首 |
24 067±2 212 |
404±82 |
4.13±0.17 |
0.028 8±0.003 4 |
508±107 |
510±101 |
99.56±0.12 |
|
表 2 不同样品浮游细菌多样性评估表
Table 2 Estimation of rhizobacteria community diversity in different samples
|
2.2.2 细菌群落组成及Beta多样性分析
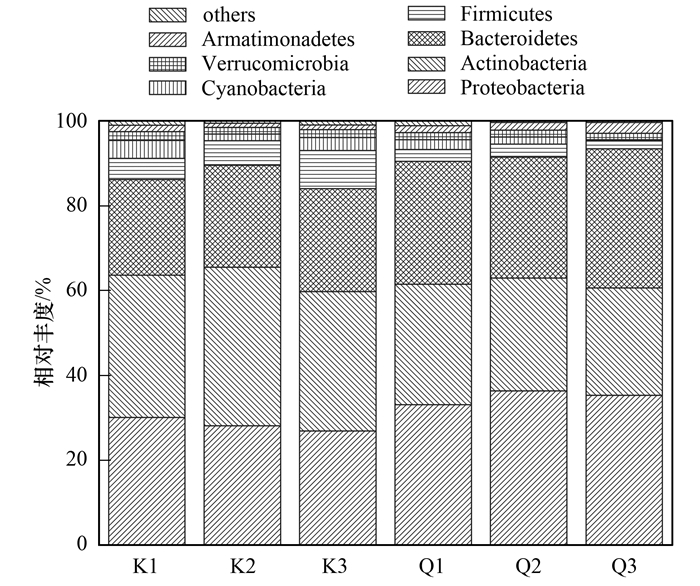
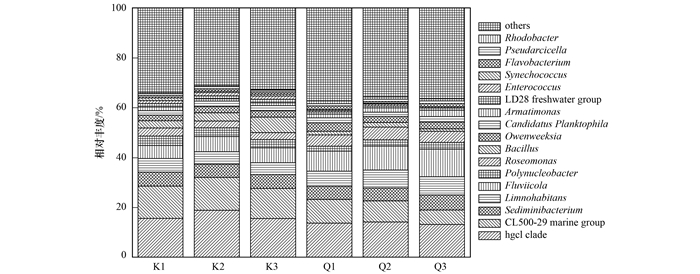
高通量测序结果表明, 库心和渠首样品浮游细菌主要由12个门组成, 包括变形菌门(Proteobacteria)、放线菌门(Actinobacteria)、拟杆菌门(Bacteroidetes)、厚壁菌门(Firmicutes)、蓝菌门(Cyanobacteria)、疣微菌门(Verrucomicrobia)等.属于变形菌门、放线菌门、拟杆菌门的序列总和占全部序列的84.10%~93.50%, 这些细菌为优势种群(图 1).对其他分类单元细菌进行分析, 库心样品包含9门、21纲、42目、61科、83属, 渠首样品包含10门、20纲、40目、56科、73属, 表现出群落组成的丰富性(图 2).
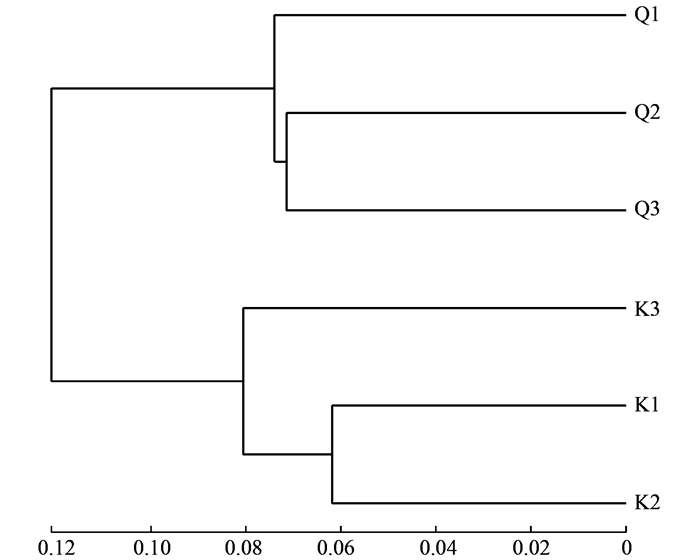
基于unweighted unifrac的UPGMA聚类分析能描述不同样品间群落差异, 如果两个样品距离较近, 表示这两个样品的物种组成较相似.丹江口库区浮游细菌群落的UPGMA聚类分析(OTUs水平)结果如图 3所示.在0.09的相似性水平上, 库心和渠首样品分为2大组, 结果表明不同生态位点样品浮游细菌群落结构有所差异.
2.2.3 不同样品差异细菌分析
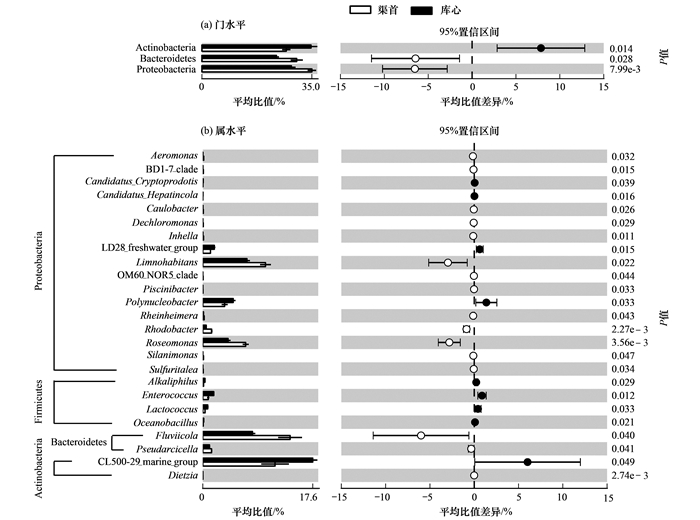
为了研究丹江口库区不同生态位点浮游细菌群落的差异, 笔者采用STAMP软件[30]在P<0.05显著性水平对门和属分类单元进行了分析, 结果如图 4所示.在门的水平上, 占群落主要组成的变形菌门、放线菌门和拟杆菌门存在显著差异[图 4(a)].其中变形菌门和拟杆菌门在渠首样品中平均占比为34.95%和30.15%, 显著高于库心样品的28.43%和23.70%.放线菌门在库心样品平均占比为34.67%, 显著高于渠首样品的26.82%.在属的水平上, 共有25个属的细菌存在差异(STAMP分析时去除unclassified的细菌), 主要分布于变形菌门(17个属)、厚壁菌门(4个属)、拟杆菌门(2个属)和放线菌门(2个属)[图 4(b)].其中库心显著高于渠首样品有9个属, 为变形菌门的Polynucleobacter[库心平均占比(K, 下同)4.85%, 渠首平均占比(Q, 下同)3.49%]、LD28 freshwater group(K:1.84%, Q:1.22%)、Candidatus_Cryptoprodotis(K:0.06%, Q:0.00%)、Candidatus_Hepatincola(K:0.02%, Q:0.00%), 厚壁菌门的Enterococcus(K:1.75%, Q:0.87%)、Lactococcus(K:0.78%, Q:0.36%)、Alkaliphilus(K: 0.34%, Q:0.11%)、Oceanobacillus(K:0. 14%, Q:0.05%)和放线菌门CL500-29 marine group(K:17.57%, Q:11.54%).渠首样品属水平显著库心样品的有16个属, 为变形菌门的Piscinibacter(K:0.00%, Q:0.02%)、OM60_NOR5_clade(K:0.00%, Q:0.03%)、Caulobacter(K:0.02%, Q: 0.07%)、Sulfuritalea(K:0.00%, Q:0.07%)、Dechloromonas(K:0.00%, Q:0.06%)、BD1-7 clade(K: 0.00%, Q:0.08%)、Silanimonas(K:0.00%, Q: 0.10%)、Inhella(K:0.01%, Q:0.12%)、Rheinheimera(K:0.10%, Q:0.22%)、Aeromonas(K:0.01%, Q:0.16%)、Rhodobacter(K:0.54%, Q:1.40%)、Roseomonas(K:4.03%, Q:6.84%)、Limnohabitans(K:7.05%, Q:10.02%), 拟杆菌门的Pseudarcicella(K:1.06%, Q:1.40%)、Fluviicola(K:7.94%, Q:13.94%)和放线菌门的Dietzia(K:0.00%, Q:0.01%).
2.3 PICRUSt功能预测分析
2.3.1 功能基因家族组成
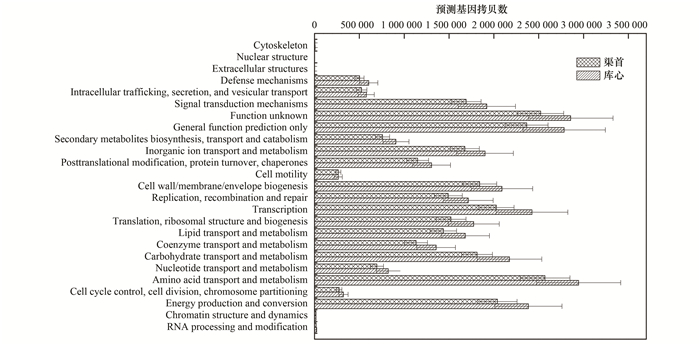
为了获得丹江口库区不同位点浮游细菌的功能, 本研究采用PICRUSt软件[18]进行菌群预测分析.基于COG(clusters of orthologs groups)数据库预测的结果表明, 除核结构功能基因家族(nuclear structure)缺失之外, 另外24个基因功能家族均具有数值(图 5).其中氨基酸运输和代谢(amino acid transport and metabolism)、转录(transcription)、能量产生和转换(energy production and conversion)、碳水化合物的运输和代谢(carbohydrate transport and metabolism)、细胞壁/细胞膜/膜结构的生物合成(cell wall/membrane/envelope biogenesis)、信号转导机制(signal transduction mechanisms)、无机离子转运与代谢(inorganic ion transport and metabolism)、翻译、核糖体结构和生物合成(translation, ribosomal structure and biogenesis)八大类功能基因家族为主要功能基因家族, 占比之和为53.55%~53.73%.未能很好注释的基本预测功能家族(general function prediction only)和未知功能基因家族(function unknown)占比分别为8.32%~8.49%和8.59%~8.93%(图 5).除胞外结构(Extracellular structures)和细胞运动(Cell motility)之外, 其他22个基因功能家族预测基因拷贝数均为库心样品高于渠首样品.
2.3.2 氮代谢相关基因分析
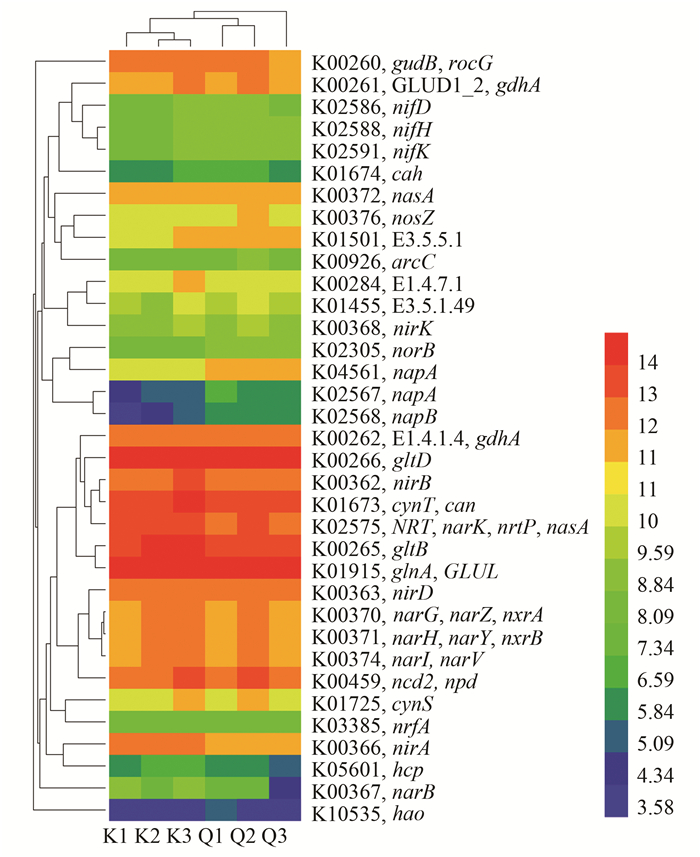
LeBrun等[31]的研究表明, PICRUSt能较为准确地预测功能基因的存在与否及其丰度, 经过比较, 准确性较微阵列分析(microarray analysis)更高.因此, PICRUSt可以用于分析丹江口库区氮循环相关基因分析.为研究丹江口库区浮游细菌氮代谢相关基因, 笔者将Kyoto Encyclopedia of Genes and Genomes(KEGG)数据库中参与氮代谢60个直系同源基因(KEGG orthology)与PICRUSt功能预测结果进行比对, 挑出浮游细菌样品中具有预测基因拷贝数的氮代谢相关的KO(表示通路), 结果如图 6所示.结果表明, KEGG数据库中60个与氮代谢相关的KO中35个KO具有数值, 表现出丰富的氮代谢能力.从图 6的氮代谢基因拷贝数聚类分析结果可以看出, 库心和渠首样品各自生物学重复(K1~3和Q1~3)聚集于一起, 表明具有较好的重复性, 库心和渠首样品在聚类分析树中相互分开, 表明不同位点样品氮代谢具有差异.其中库心高于渠首的KO有20个, 渠首高于库心的KO有15个(图 6).
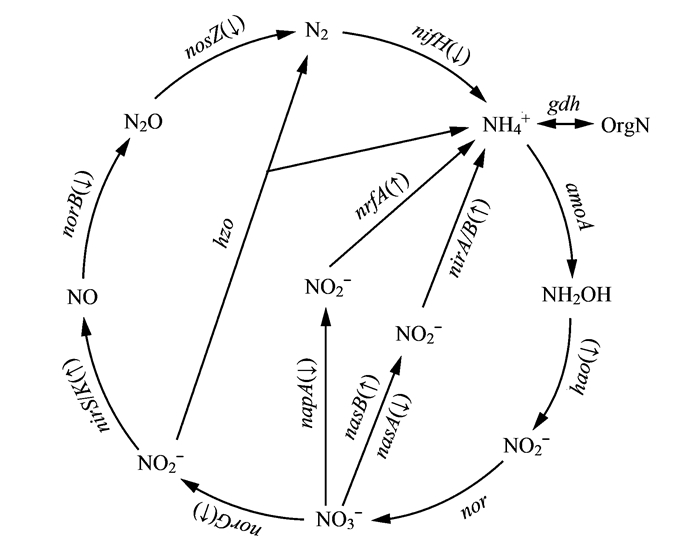
微生物介导的氮代谢是地球氮循环的主要驱动力, 本研究重点比较分析不同位点浮游细菌氮循环(硝化、反硝化、氨化及固氮等过程)相关基因表达差异.因此, 将35个氮代谢相关KO中参与氮循环的关键基因筛选出, 并进行统计归类, 得到氮循环途径各类基因数量(图 7)[7].结果表明, PICRUSt预测了部分氮循环关键基因的相对丰度, 主要有固氮作用的固氮酶基因nifH, 硝化过程的羟胺氧化还原酶基因hao, 反硝化过程的硝酸还原酶基因narG、亚硝酸还原酶基因nirK、一氧化氮还原酶基因norB以及氧化亚氮还原酶的编码基因nosZ, 氮同化还原及异化还原作用过程中的硝酸盐同化还原酶基因nasA、narB和异化还原酶基因napA、亚硝酸盐同化还原酶基因nirA、nirB和异化还原酶基因nrfA[7, 32].其中库心样品基因相对丰度高于渠首样品的有氮同化还原及异化还原作用过程中的nirA、nirB、narB和nrfA, 反硝化过程的narG, 硝化过程的nirK .渠首样品基因相对丰度高于库心样品的有固氮作用nifH, 硝化过程的hao, 氮同化还原及异化还原作用过程中的napA和nasA, 反硝化过程的nosZ和norB(图 7).
3 讨论
3.1 丹江口库区表层浮游细菌群落分析
浮游细菌是水生态系统的重要组成部分, 研究其群落组成和功能对于管理和维护丹江口水库生态环境具有深远的意义[3].长期监测表明丹江口库区承载着较大的氮、磷负荷, 其中TN明显超标[4, 5]. 2016年5月监测结果与上述结果一致, 库心、渠首样品TN均超过0.90 mg ·L-1, 为Ⅲ类地表水标准.高通量测序结果表明, 丹江口库区库心和渠首样品具有丰富的群落组成, 由变形菌门、放线菌门、拟杆菌门、厚壁菌门等12个门组成, 渠首样品浮游细菌群落多样性高于库心样品.微生物介导的氮代谢是地球氮循环的主要驱动力, 之前丹江口库区浮游细菌群落分析表明浮游细菌受到水体中TN、NH4+-N影响[5].库心和渠首样品门和属的水平上差异分析表明, 显著差异的细菌主要分布于变形菌门、放线菌门、拟杆菌门和厚壁菌门.之前研究表明, 水体中常见固氮菌Clusters I类群由大部分α-、β-、γ-、δ-变形菌和蓝细菌/蓝藻(Cyanobacteria)等组成, 同时氨氧化细菌(ammonia oxidizing bacteria, AOB)主要包括β-变形菌纲的亚硝化单胞菌(Nitrosomonas)与亚硝化螺菌属(Nitrosospira)以及γ-变形菌纲的亚硝化球菌属(Nitrosococcus)等组成[7].属水平上, 变形菌门的Rheinheimera、Polynucleobacter、Dechloromonas、Rhodobacter、Limnohabitans、Rheinheimera, 拟杆菌门的Pseudarcicella、Fluviicola和放线菌门的Dietzia等25个属的细菌存在显著差异(图 4).这些属的细菌中, Rheinheimera在氮的固定[33]、Polynucleobacter在溶解性有机物降解[34]、Dechloromonas在亚硝酸盐还原为一氧化氮[35]、Rhodobacter在有机氮复合物降解[36]、Fluviicola在溶解性有机氮利用[37]、Limnohabitans在硝酸盐还原[38]等氮的循环过程起到作用, 影响水体中氮元素生物化学地球循环.
3.2 丹江口库区浮游细菌功能预测分析
微生物可以通过氨化作用、硝化作用、反硝化作用、固氮作用等将N2、无机氮化合物、有机氮化合物在自然界中相互转化, 从而驱动地球氮元素的生物地球化学循环[6, 7].丹江口库区面临TN超标的现状, 了解丹江口库区氮循环过程中细菌群落组成和功能对丹江口水质保护具有重要意义. Dang等[12]采用结合荧光定量PCR和高通量测序技术测定了丹江口库区表层沉积物中氨氧化细菌和氨氧化古菌基因丰度和分布.针对丹江口库区浮游细菌氮循环过程中细菌群落组成和功能研究较少开展的现状, 基于高通量测序的PICRUSt分析能很好满足研究的需要[31].董志颖等[19]采用PICRUSt功能预测分析过量氮输入对细菌群落代谢潜力的影响, 发现氮输入在一定程度上影响了微宇宙中的固氮、硝化、异化硝酸盐还原、反硝化、异化硝酸盐还原到铵和同化硝酸盐还原途径的关键基因.本论文对库心、渠首高通量测序结果进行PICRUSt功能预测分析, 结果表明丹江口库区浮游细菌具有丰富的功能, 其中22个基因功能家族预测基因拷贝数均为库心样品高于渠首样品, 该结果与浮游细菌群落多样性分析相反.氮代谢相关的KO分析表明, 库心高于渠首的KO有20个, 渠首高于库心的KO有15个.接着对氮循环(硝化、反硝化、氨化及固氮等过程)基因进行分析, 结果检测到涉及固氮作用(nifH)、硝化作用(hao)、反硝化作用(narG、nirK等)和氮同化还原及异化还原作用(nasA、narB等)等基因的相对丰度, 这些基因在氮循环中起到关键作用[7, 32].对不同位点氮循环关键基因相对丰度进行比较分析, 发现库心高于渠首样品的有6个, 主要涉及氮同化还原及异化还原作用(nirA、nirB、narB和nrfA, )、反硝化作用(narG)和硝化作用(narG); 渠首高于库心样品的有6个, 主要涉及固氮作用(nifH)、硝化作用(hao)反硝化(nosZ, norB)氮同化还原及异化还原作用(napA、nasA).结合基因功能家族预测基因拷贝数和氮循环相关基因丰度分析, 丹江口库区库浮游细菌氮代谢能力整体趋势为库心高于渠首.高通量测序技术结合PICRUSt功能预测分析在丹江口库区浮游细菌研究中应用, 为探明库区浮游细菌群落组成和功能提供重要帮助.
4 结论
(1) 本研究采用高通量测序的方法研究了丹江口库区库心和渠首两个生态位点浮游细菌群落组成, 发现其主要由变形菌门、放线菌门、拟杆菌门、厚壁菌门等12个门139属细菌组成.细菌群落分析表明不同位点群落组成有差异, 在门的水平上变形菌门、放线菌门和拟杆菌门存在显著差异; 在属的水平上, 共有Rheinheimera、Polynucleobacter、Dechloromonas、Pseudarcicella、Fluviicola等25个属存在显著差异.渠首样品浮游细菌群落多样性高于库心样品.
(2) PICRUSt功能预测分析表明, 浮游细菌涉及氨基酸运输和代谢、转录等24个基因功能家族, 表现出功能上的丰富性.结合基因功能家族预测基因拷贝数和氮循环相关基因丰度分析, 丹江口库区浮游细菌氮代谢能力整体趋势为库心高于渠首.
 2019, Vol. 40
2019, Vol. 40