 2017, Vol. 38
2017, Vol. 38



2. 启源(西安)大荣环保科技有限公司, 西安 710018
2. Qiyuan(Xi'an) Dae Young Environmental Protection Technology Co., Ltd., Xi'an 710018, China
氨选择催化还原NOx(NH3-SCR)技术因其转化率高、选择性好、实用性强等优点得到广泛的工业化应用,但该技术存在明显的缺点:① NH3是一种有毒腐蚀性气体,存储和输运过程中有一定的危险,对管路设备要求高,造价昂贵;② 该过程NH3需要计量控制加入量,容易泄漏或反应不完全而造成二次污染;③ NH3与烟道气中的SO2反应,形成腐蚀性的NH4HSO4,易使催化剂中毒;④ 传统的V-W-Ti催化剂成本高、操作温度高以及钒的本征毒性[1].烃类催化还原NOx(HC-SCR)作为极具应用潜力的脱硝技术受到广泛关注[2].相比其他烃类还原剂,甲烷是天然气中含量最多的组分,来源丰富,价格低廉,因此甲烷催化还原NOx(CH4-SCR)技术成为研究热点[3~5].然而由于较高的C—H键能(435 kJ·mol-1)造成CH4在催化还原NOx中难以活化.
已报道的CH4-SCR脱硝催化剂中,Co基/分子筛催化剂因其活性和N2选择性高得到了广泛研究. Co基/分子筛催化剂在CH4-SCR中的研究主要集中在5个方面:① Co基/分子筛催化剂制备方法的优化设计,如相比浸渍法,离子交换法制备的Co/MOR催化剂在CH4-SCR中具有更高的催化活性[6]. ② 助剂对Co基/分子筛催化剂活性的促进,如In-Co/ZSM-5[3]、Co-Ni/MOR[7]和Co-Zn/BZZ[8]等. ③ Co基/分子筛催化剂载体的优化选择,如相比FER和BEA分子筛载体,Co-Pd/MOR和Co-Pd/ZSM-5催化剂具有较强的催化活性[9]. ④ Co基/分子筛催化剂的抗硫耐水性, 如张金桥等[10]认为由于部分活性位被含硫化合物覆盖而导致吸附的NO和活性中间产物容量的下降,从而降低CH4-SCR反应活性. ⑤ 催化反应机制[11].在以上5个方面的研究中,助剂和载体是催化剂的重要组成部分,是影响催化剂活性的重要因素,然而目前的相关研究仅局限于助剂或载体的优化,同时优化Co基/分子筛催化剂的助剂和载体的研究鲜有报道.
过渡金属具有可变的化学价态,可作为具有氧化还原活性位.微孔分子筛具有丰富的酸性位,可作为脱硝催化剂的载体.因此本文采用过渡金属(Mn、Fe和Zn)为助剂,典型微孔分子筛(ZSM-5、SAPO-34和Beta)为载体,通过同时优化助剂和载体,制备高活性的Co基/分子筛催化剂,并探明Co基分子筛催化剂的构效关系及催化反应机制.
1 材料与方法 1.1 催化剂制备H-ZSM-5(硅铝比=25)、H-SAPO-34(硅铝比=1) 和H-Beta(硅铝比=25) 等氢型分子筛购自南开大学催化剂厂.双组分过渡金属(Mn、Fe或Zn)-Co/分子筛催化剂采用共浸渍法制备.将分子筛浸入适量的浓度为0.1mol·L-1的Co(NO3)2和过渡金属硝酸盐的混合溶液中,室温下放置24 h后,于120℃下干燥8 h,500℃下焙烧2 h,制得一系列双组分过渡金属-Co/分子筛催化剂.使用美国PerkinElmer公司的Optima 7000DV型电感耦合等离子体发射光谱仪(ICP-OES)测试样品中的金属含量,制得催化剂中金属的质量分数分别为2.04%Co-1.97%Mn/ZSM-5、1.96%Co-1.95%Fe/ZSM-5、2.03%Co-2.05%Zn/ZSM-5、2.05%Co/ZSM-5、2.01%Co-1.92%Fe/SAPO-34、1.95%Co-2.04%Fe/Beta.所有催化剂均经过研磨、压片、过筛,制成40~60目的颗粒用于活性测试.
1.2 催化剂的活性评价催化活性测试在长度为30 cm,内径为10 mm微型固定床催化反应装置中进行,实验模拟气各组分的体积分数分别为φ(CH4)=1×10-3、φ(NO)=6×10-4、φ(O2)=5×10-2和N2为载气.反应气体(CH4、NO、O2和N2)经质量流量计控制,通过缓冲瓶混合后进入催化反应器.实验温度范围为350~550℃.催化剂装量2 mL,气体总流量为500 mL·min-1,空速15 000 h-1,反应物和产物中的NO、NO2的浓度由NO/NO2分析仪测量,N2O浓度采用配有热导检测器的气相色谱(灵华GC9890) 测量,其中porapak Q色谱柱用于分离产物中N2O、CO2和NO.催化剂活性分别用NOx转化率、CO和CO2产率表示:
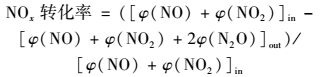
|
(1) |
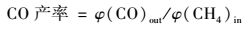
|
(2) |
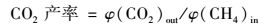
|
(3) |
式中,[φ(NO)+φ(NO2)]in为进气中NO和NO2的体积分数之和;[φ(NO)+φ(NO2)+2φ(N2O)]out为出口NO、NO2和N2O的体积分数之和;φ(CO)out为出口CO的体积分数;φ(CO)out为出口CO2的体积分数;φ(CH4)in为进气中CH4的体积分数.
1.3 催化剂表征XRD衍射用阶梯扫描方式.在Rigaku D/Max ⅢB全自动X射线粉末衍射仪上采集,Cu/Kα辐射源,功率40 kV×40 mA,衍射束置石墨单色器除去Kβ辐射,入射波长为0.154 05 nm,阶宽0.02°,2θ为5°~80°,每步计数时间4 s.
采用XPS分析样品表面的各元素相对含量比例.仪器为美国PHI公司的PHI 5000C ESCA System(经过美国RBD公司升级),采用条件为铝/镁靶,高压14.0 kV, 功率250 W, 真空优于1.33×10-6 Pa.采用美国RBD公司的RBD147数据采集卡和AugerScan3.21软件分别采集样品的0~1 200(1000) eV的全扫描谱(通能为93.9 eV),而后采集各元素相关轨道的窄扫描谱(通能为23.5 eV或46.95 eV).以C1s=284.8 eV为基准进行结合能校正,采用XPSPeak4.1软件进行分峰拟合.
原位漫反射红外光谱(DRIFT)在配有原位漫反射附件的德国Bruker公司Vertex 70型傅里叶变换红外光谱仪上进行,使用液氮冷却的MCT检测器, 扫描范围1 000~4 000 cm-1,分辨率4 cm-1,扫描次数100次.样品粉末先在450℃下使用N2吹扫45 min,而后降温至300℃,采集背景,然后通入=6×10-4φ(NO)/5×10-2φ(O2)/N2、或1×10-3φ(CH4)/5×10-2φ(O2)/N2、或6×10-4φ(NO)/1×10-3φ(CH4)/5×10-2φ(O2)/N2,12 min后,用N2吹扫10 min再采集光谱.
2 结果与讨论 2.1 助剂和载体优化图 1(a)考察了3种助剂(Mn、Fe和Zn)对双组分Co-M/ZSM-5催化剂活性的影响.在3种催化剂上,NOx转化率随温度的变化曲线呈典型的“火山型”趋势:即初始NOx转化率随温度升高逐渐增加,当达到最佳工作温度后,NOx转化率随温度升高而逐渐减小. Co-Fe/ZSM-5、Co-Mn/ZSM-5和Co/ZSM-5催化剂的最佳活性温度均为450℃,NOx最高转化率分别为46.4%、34.5%和36.7%. Co-Zn/ZSM-5的最佳活性温度则往高温段偏移了约50℃,在500℃时NOx最高转化率为40.3%.当温度低于450℃时,Co-Fe/ZSM-5表现出最好的低温催化活性;当温度高于450℃时,Co-Zn/ZSM-5显示出较好的高温催化活性.有研究表明,Fe作为助剂提高Co/ZSM-5低温催化活性是由于在低温时Fe具有较强的催化氧化NO(NO+O2NO2)能力[12],NO氧化被普遍认为是NOx催化还原的初始步骤[13],故Co-Fe/ZSM-5具有较强的低温催化活性. Co-Zn/ZSM-5具有较好的高温催化活性则是由于在高温时Zn可抑制甲烷的催化燃烧(CH4+O2CO2),从而提高了Co-Zn/ZSM-5的高温催化活性[14].

|
图 1 助剂对Co/ZSM-5催化活性的影响 Fig. 1 Effect of promoters on the activity of Co/ZSM-5 catalyst |
图 1(b)和1(c)考察了3种助剂(Mn、Fe和Zn)对双组分Co-M/ZSM-5催化剂CO和CO2的产率的影响.在图 1(b)中,3种催化剂上CO产率随温度升高逐渐增加,当达到一定温度时,CO产率随温度升高而逐渐减小. Co-Fe/ZSM-5、Co-Mn/ZSM-5和Co/ZSM-5催化剂上CO产率达到最大时的温度分别为400、400和450℃.其中Co-Fe/ZSM-5和Co-Mn/ZSM-5的CO产率变化相似,且最大CO产率小于Co/ZSM-5.而Co-Zn/ZSM-5的最大CO产率则大于Co/ZSM-5.在图 1(c)中,3种催化剂上CO2产率均随温度升高逐渐增加,其中Co-Fe/ZSM-5和Co-Mn/ZSM-5的CO2产率基本相似,均大于Co/ZSM-5,而Co-Zn/ZSM-5的CO2产率则小于Co/ZSM-5.可见Fe和Mn可提高CH4转化成CO2选择性,Zn则可抑制甲烷的催化燃烧.在烃类选择催化还原NOx中,CO2的选择性高则意味着有毒气体CO的产率低,因此,综合NOx转化率、CO和CO2产率,笔者认为Fe可作为合适的助剂提高Co基分子筛的催化活性.
为进一步提高Co-Fe/分子筛的催化活性,笔者对3种典型微孔分子筛(ZSM-5、SAPO-34和Beta)进行了载体优化(见图 2).在图 2(a)中,3种Co-Fe/分子筛催化剂上NOx转化率随温度的变化曲线也呈典型的“火山型”.在整个温度区间内,催化活性顺序依次为Co-Fe/SAPO-34>Co-Fe/ZSM-5>Co-Fe/Beta,且3种Co-Fe/分子筛催化剂的最佳活性温度均在450℃.其中活性最高的Co-Fe/SAPO-34催化剂上NOx最大转化效率达到52.7%.载体的不同会造成活性组分形态和分布的差异,从而导致催化活性的差别. Pieterse等[9]通过H2-TPR探究了Co-Pd/FER和Co-Pd/MOR脱硝催化剂中钴物种的还原性差异.本课题组[15]采用XPS探明了Fe2+和Fe3+物种在Fe/分子筛(ZSM-5、Beta、MOR和FER)脱硝催化剂中含量的差异.因此,分析Co-Fe/分子筛催化剂中钴和铁物种的形态差异,及其与催化活性间的构效关系具有重要意义(见2.3节).

|
图 2 载体对Co-Fe/分子筛催化活性的影响 Fig. 2 Effect of supports on the activity of Co-Fe/zeolites catalysts |
图 2(b)和2(c)考察了载体对Co-Fe/分子筛催化剂CO和CO2的产率影响.在图 2(b)中,3种催化剂上CO产率达到最大时的温度均为分别为400℃.其中Co-Fe/SAPO-34的CO产率最小,Co-Fe/Beta的CO产率最大.在图 2(c)中,3种催化剂上CO2产率均随温度升高逐渐增加,CO2产率的顺序依次为Co-Fe/SAPO-34>Co-Fe/ZSM-5>Co-Fe/Beta.可见SAPO-34可作为合适的载体提高Co基分子筛的催化活性.
2.2 催化剂稳定性图 3考察了450℃时3种Co-Fe/分子筛催化剂在SO2、H2O和CO2等污染组分下的稳定性.当原料气中添加体积分数为15% CO2时,3种Co-Fe/分子筛催化剂的活性变化不大,这说明CO2不会抑制Co-Fe/分子筛催化剂活性.当在含有15%CO2的原料气中通入φ(H2O)10%时,3种Co-Fe/分子筛催化剂的活性均略有下降,但原料气去掉H2O后,3种Co-Fe/分子筛的活性基本恢复到初始水平,因此,H2O对Co-Fe/分子筛催化剂活性的抑制是可逆的.当φ(SO2)=2×10-4通入到原料气时,3种Co-Fe/分子筛催化剂的活性明显下降,且同时通入φ(CO2)15%、φ(H2O)10%和φ(SO2)=2×10-4时,3种Co-Fe/分子筛催化剂的活性继续下降,Co-Fe/SAPO-34、Co-Fe/ZSM-5和Co-Fe/Beta的活性分别下降到36.2%、30.5%和26.3%.当原料气中不通入H2O、SO2和CO2后,3种Co-Fe/分子筛催化剂的活性部分恢复,说明SO2对Co-Fe/分子筛催化剂的活性抑制是部分可逆.

|
图 3 Co-Fe/分子筛的稳定性 Fig. 3 Stability of Co-Fe/zeolite catalysts |
图 4为3种Co-Fe/分子筛催化剂的XRD谱图.在Co-Fe/ZSM-5和Co-Fe/Beta中有微弱的α-Fe2O3的衍射峰(2θ为33.1°和35.6°)[15], 而Co-Fe/SAPO-34中未发现α-Fe2O3的衍射峰,这表明Co-Fe/分子筛催化剂中铁物相的尺寸太小(<3~5 nm)且高度分散在催化剂中造成XRD手段无法检测[16]. Co物种在3种Co-Fe/分子筛催化剂中均以Co3O4(ICDD#01-080-1540)、CoO(ICDD#01-071-1178)、Co(OH)2(ICDD#00-001-0357) 和CoAl2O4(ICDD#01-082-2251) 形式存在.文献[11]报道了CoO和Co(OH)2是Co-基/分子筛的主要活性位.笔者通过XPS进一步分析了3种催化剂表面的Co和Fe的物种形态.

|
图 4 Co-Fe/分子筛催化剂的XRD谱图 Fig. 4 XRD patterns of Co-Fe/zeolites catalysts |
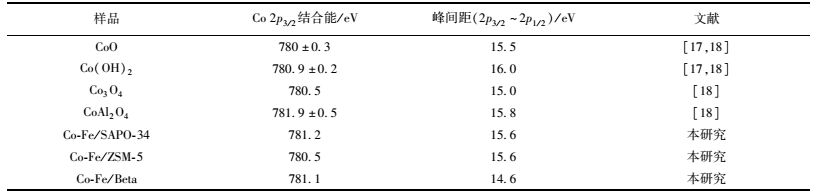
图 5为3种Co-Fe/分子筛催化剂的Co 2p谱图.从中可见,Co-Fe/SAPO-34、Co-Fe/ZSM-5和Co-Fe/Beta催化剂的Co 2p3/2结合能分别为781.4、780.5和780.8 eV,峰间距(Co 2p1/2~Co 2p3/2)分别为15.8、15.5和14.6 eV.参考表 1罗列的相关Co化合物的相应参考数值,Co-Fe/SAPO-34催化剂表面以CoO和Co(OH)2为主,Co-Fe/ZSM-5催化剂表面则以Co3O4和Co(OH)2为主.然而,Co-Fe/Beta催化剂的峰间距(Co 2p1/2~Co 2p3/2)与表 2中Co的有关化合物的相应参考数值相差较大,根据其Co 2p3/2结合能判断可能以CoO、CoAl2O4和Co3O4为主. Lónyi等[11]认为Co/分子筛催化剂在CH4-SCR反应中具有较强的活化活性是由于NO和NO2在Co2+和[Co—OH]+等活性位上可转化成有利于反应的NO3-/NO+离子对中间产物.因此不同催化剂中Co物种的差异是造成催化活性Co-Fe/SAPO-34>Co-Fe/ZSM-5>Co-Fe/Beta催化剂的原因之一.

|
图 5 Co-Fe/分子筛催化剂上Co 2p谱图 Fig. 5 Co 2p-XPS spectra of Co-Fe/zeolites catalysts |
|
|
表 1 Co 2p的XPS参数及实验数据列表 Table 1 XPS parameters from Co 2p in Co-Fe/zeolites and relevant reference compounds |
|
|
表 2 Co-Fe/分子筛催化剂的Fe 2p3/2的XPS结果 Table 2 XPS Parameters from Fe 2p3/2 in Co-Fe/zeolites |

为了阐明载体对催化剂中Fe物种化学形态分布的影响,图 6采用了Gaussian-Lorentzian模型对3种Co-Fe/分子筛催化剂的Fe 2p3/2谱图进行了分峰.文献[19~21]报道了Fe2+和Fe3+的峰位置分别为709.5~711 eV和712~713 eV.这表明3种Co-Fe/分子筛催化剂表面均含有Fe2+和Fe3+.但3种Co-Fe/分子筛催化剂中Fe2+和Fe3+峰的结合能和面积(即Fe2+和Fe3+含量)有较大的差异.相比Co-Fe/SAPO-34催化剂,Co-Fe/Beta的Fe2+和Fe3+峰的结合能明显向低的结合能方向移动. Co-Fe/ZSM-5的Fe2+和Fe3+峰的结合能位置虽与Co-Fe/SAPO-34催化剂的相别不大,但两种催化剂表面的c(Fe2+)/c(Fe3+)存在明显不同,Co-Fe/ZSM-5催化剂表面Fe2+含量明显大于Fe3+含量,而Co-Fe/SAPO-34催化剂则相反. 表 2罗列了3种催化剂的Fe 2p3/2分峰后的相关结果.从表 2中可见,

|
图 6 Co-Fe/分子筛催化剂上Fe 2p3/2谱图 Fig. 6 Fe(2p3/2)-XPS spectra of Co-Fe/zeolites catalysts |
3种Co-Fe/分子筛催化剂表面Fe2+/Fe3+比值依次为Co-Fe/ZSM-5>Co-Fe/SAPO-34>Co-Fe/Beta. Fe2+和Fe3+均认为是烃类催化还原NOx的活性位[22]. Lobree等[23]提出了Fe/ZSM-5催化剂上烃类催化还原NOx的机制:首先Fe2+与O2反应生成Fe3+(O2-),然后Fe3+(O2-)与NO反应生成Fe3+(O-)(NO2)或Fe3+(NO3-),当这些生成的亚硝酸盐或硝酸盐与烃类反应会产生CN和NCO等中间产物,最后CN和NCO等中间产物分解,或与NO2或O2反应生成N2,同时Fe3+还原成Fe2+.结合图 2的活性评价结果,即Co-Fe/SAPO-34具有最好的催化活性,笔者认为合适的c(Fe2+)/c(Fe3+)比对于烃类催化还原NOx至关重要,过低的c(Fe2+)/c(Fe3+)比不利于亚硝酸盐或硝酸盐的形成,过高的c(Fe2+)/c(Fe3+)比则不利于CN和NCO等中间产物分解及其与NO2或O2反应生成N2.
2.4 反应机制图 7为NO在3种Co-Fe/分子筛催化剂中吸附氧化的原位-DRIFT谱图. 3 250 cm-1处的吸收峰归属于Brønsted酸的羟基(V—OH)[24],从峰强度得到Co-Fe/SAPO-34的Brønsted酸强度大于其他2种催化剂.在3种催化剂上,随着温度的升高,Brønsted酸强度均显著减少. Brønsted酸对于中间产物(NO3-物种)的生成至关重要,因为NO在Brønsted酸上的吸附并生成NO+被认为是HC-SCR的初始步骤[3, 25]. 2 348 cm-1和2 351 cm-1处的吸收峰为气态NO2的卫星峰.文献[26]通过方程(4) 揭示了这些物种的反应过程:

|
图 7 NO在不同催化剂上的吸附氧化的原位-DRIFT谱图 Fig. 7 DRIFT spectra of NO adsorption and oxidation on various catalysts at temperature from 300-550℃ |

|
(4) |
式中,Z-代表带一个负电荷的分子筛骨架. 1 100~1 650 cm-1范围内的吸收峰归属于不同类型的表面硝酸盐[27].由活性位上NO+生成的桥接硝酸盐(1 650 cm-1和1 649 cm-1)、双齿硝酸盐(1 558、1 339、1 065和1 058 cm-1)和单齿硝酸盐(1 536 cm-1)在不同催化剂和不同温度中出现,如Co-Fe/SAPO-34催化剂上的桥接硝酸盐(1 650 cm-1)和双齿硝酸盐(1 558 cm-1)在反应温度为300℃时生成,而Co-Fe/Beta催化剂上的桥接硝酸盐(1 649 cm-1)和双齿硝酸盐(1 558 cm-1)则在反应温度为450℃时生成.这表明由于3种催化剂中Co和Fe的活性位物种的不同,导致反应中间产物硝酸盐生成温度的不同,从而导致其活性的差异.
CH4在3种催化剂中吸附氧化的原位-DRIFT结果见图 8.不同于NO吸附氧化的原位-DRIFT结果,Brønsted酸强度未随温度的升高而逐渐减小,其中在350℃时强度最大,而在450℃强度最小.强甲烷吸收峰出现在3 010 cm-1[28],且其强度随着温度升高而逐渐减小. CO2的吸收峰出现在2 348 cm-1和2 354 cm-1[26],且由于温度升高造成CO2的脱附,使得其吸收峰强度随着温度升高逐渐减少. —COO吸收峰(1 542 cm-1和1 454 cm-1)[29]和H—COO吸收峰(1 518 cm-1)[29]随着温度的升高而逐渐增大.以上反应物种的吸收峰强度的变化表明:CH4在300℃时吸附在Brønsted酸上使得V—OH强度减小,在350℃脱附造成Brønsted酸的V—OH强度增大,当温度升高到450℃时,CH4开始活化,在Brønsted酸上生成—COO和H—COO等物种造成Brønsted酸上的V—OH强度减少,当温度达到550℃时,因生成的—COO和H—COO等物种氧化成CO2并从Brønsted酸脱附,从而造成Brønsted酸的V—OH吸收峰强度增大.在Co-Fe/SAPO-34催化剂上,—COO吸收峰(1 542 cm-1)在300℃时出现,而Co-Fe/ZSM-5和Co-Fe/Beta催化剂中—COO吸收峰(1 542 cm-1)和H—COO吸收峰(1 518 cm-1)在温度高于350℃时出现,这表明由于3种催化剂活性位的不同,导致CH4在3种催化剂上的活化程度的差异,其中在Co-Fe/SAPO-34催化剂上,CH4在低温时易活化.

|
图 8 CH4在不同催化剂上的吸附氧化的原位-DRIFT谱图 Fig. 8 DRIFT spectra of CH4 adsorption and oxidation on various catalysts at temperature from 300-550℃ |
如图 9所示,3种催化剂上CH4-SCR反应的原位-DRIFT谱图. Brønsted酸的V—OH(3 250 cm-1)出现在高温段(450~550℃).这表明在低温时(300~350℃), NO和CH4吸附在Brønsted酸造成低强度的V—OH吸收峰,而在高温时(450~550℃),由于吸附的NO和CH4在活性位上生成中间产物并转化成N2和CO2,从而使得V—OH吸收峰强度增大.其中最强的V—OH吸收峰出现在450℃,这意味着中间产物最大限度地转化成产物N2和CO2.吸收峰1 691 cm-1和1 535~1 541 cm-1可分别归属于—CO[30]和—COO[29]. 1 022~1 036 cm-1和1 069 cm-1吸收峰则分别归属于碳酸氢盐和双齿硝酸盐[31]. CO2吸收峰则出现在2 347~2 351 cm-1处.同样,3种催化剂中不同反应中间产物在不同温度的吸收峰强度有较大差异.如相对其他催化剂,Co-Fe/SAPO中—CO(1 691 cm-1)和—COO(1 535 cm-1)在300℃具有较强的吸收峰强度,表明Co-Fe/SAPO催化剂中CH4在低温时易活化,故Co-Fe/SAPO具有较强的低温活性.而在高温时(550℃),Co-Fe/ZSM-5催化剂中碳酸氢盐吸收峰强度较高,这表明在高温时CH4的燃烧反应为主要反应,从而导致Co-Fe/ZSM-5催化剂的高温活性较差.

|
图 9 不同催化剂上CH4-SCR反应的原位-DRIFT谱图 Fig. 9 DRIFT spectra of CH4-SCR on various catalysts at temperature from 300-550℃ |
综合以上原位-DRIFT结果,Co-Fe/分子筛催化剂上CH4-SCR反应的机制见图 10.首先NO吸附在Brønsted酸上并生成NO+,同时CH4在Brønsted酸位上吸附并活化成—CO和—COO等活性反应物种;然后NO+在Co2+和Fe3+等催化活性位上转化成反应中间产物硝酸盐等;最后硝酸盐与活化物种(—CO和—COO)反应生成N2和CO2.

|
图 10 Co-Fe/分子筛催化剂上CH4-SCR反应机制 Fig. 10 Reaction mechanism of CH4-SCR over Co-Fe/zeolite catalysts |
(1) 在CH4-SCR中,过渡金属Fe作为助剂和SAPO-34分子筛作为载体优化设计的Co-Fe/SAPO-34催化剂具有最高的催化活性,NOx转化率在450℃时可达56.5%. CO2对Co-Fe/分子筛催化剂活性无明显抑制作用,H2O对催化剂的活性抑制是可逆的,同时通入H2O和SO2会加重催化活性的抑制作用.
(2) Co和Fe物种在不同的Co-Fe/分子筛催化剂中存在明显的差异,从而造成不同的Co-Fe/分子筛催化剂活性的不同. Co-Fe/SAPO-34催化剂表面以CoO和Co(OH)2为主,Co-Fe/ZSM-5催化剂表面则以Co3O4和Co(OH)2为主, Co-Fe/Beta催化剂则可能以CoO、CoAl2O4和Co3O4为主. Fe2+/Fe3+含量比依次为Co-Fe/ZSM-5>Co-Fe/SAPO-34>Co-Fe/Beta.活性组分Co物种的形态和合适的c(Fe2+)/c(Fe3+)比对于Co-Fe/分子筛催化剂上甲烷催化还原NOx至关重要.
(3) Co-Fe/分子筛催化剂上CH4-SCR的反应机制为Brønsted酸上吸附氧化生成NO+,同时CH4在Brønsted酸位上吸附活化成—CO和—COO等活性物种,然后NO+在Co2+和Fe3+等催化活性位上转化成硝酸盐等中间产物,最后硝酸盐与活化物种(—CO和—COO)反应生成N2和CO2.
| [1] | 徐彬, 陈天虎, 刘海波, 等. 热处理天然褐铁矿制备γ-Fe2O3及其NH3-SCR活性探究[J]. 环境科学, 2016, 37(7): 2807–2814. Xu B, Chen T H, Liu H B, et al. Preparation of γ-Fe2O3 catalyst by heat treatment of natural limonite for selective catalytic reduction of NO by NH3[J]. Environmental Science, 2016, 37(7): 2807–2814. |
| [2] | Bartolomeu R, Mendes A N, Fernandes A, et al. NOx SCR with decane using Ag-MFI catalysts:on the effect of silver content and co-cation presence[J]. Catalysis Science & Technology, 2016, 6(9): 3038–3048. |
| [3] | Lónyi F, Solt H E, Valyon J, et al. The activation of NO and CH4 for NO-SCR reaction over In-and Co-containing H-ZSM-5 catalysts[J]. Journal of Molecular Catalysis A:Chemical, 2011, 345(1-2): 75–80. DOI: 10.1016/j.molcata.2011.05.021 |
| [4] | Mendes A N, Zholobenko V L, Thibault-Starzyk F, et al. On the enhancing effect of Ce in Pd-MOR catalysts for NOx CH4-SCR:a structure-reactivity study[J]. Applied Catalysis B:Environmental, 2016, 195: 121–131. DOI: 10.1016/j.apcatb.2016.05.004 |
| [5] | Lee K J, Rao K N, Yu C Y, et al. Synthesis and characterisation of K-Ag/Al2O3 catalysts for CH4-SCR of NOx:effect of SO2[J]. Research on Chemical Intermediates, 2013, 39(3): 1463–1479. DOI: 10.1007/s11164-012-0704-9 |
| [6] | 李滨, 王虹, 丁福臣, 等. 制备方法对Co-MOR催化剂CH4选择还原NO性能的影响[J]. 物理化学学报, 2013, 29(6): 1289–1296. Li B, Wang H, Ding F C, et al. Effects of preparation methods on the catalytic performance of selective catalytic reduction of NO with CH4 over Co-MOR catalysts[J]. Acta Physico-Chimica Sinica, 2013, 29(6): 1289–1296. |
| [7] | Campa M C, Pietrogiacomi D, Occhiuzzi M. The simultaneous selective catalytic reduction of N2O and NOx with CH4 on Co-and Ni-exchanged mordenite[J]. Applied Catalysis B:Environmental, 2015, 168-169: 293–302. DOI: 10.1016/j.apcatb.2014.12.040 |
| [8] | Chen J Q, Chen S W, Li R F, et al. Effect of transition metal additives on composite cobalt catalyst for NOx reduction with CH4[J]. Russian Journal of Physical Chemistry A, 2014, 88(7): 1103–1112. DOI: 10.1134/S0036024414070206 |
| [9] | Pieterse J A Z, van den Brink R W, Booneveld S, et al. Influence of zeolite structure on the activity and durability of Co-Pd-zeolite catalysts in the reduction of NOx with methane[J]. Applied Catalysis B:Environmental, 2003, 46(2): 239–250. DOI: 10.1016/S0926-3373(03)00213-3 |
| [10] | 张金桥, 刘于英, 贺勇, 等. SO2对CoH-ZSM-5催化CH4还原NO催化性能的影响[J]. 环境科学, 2006, 27(9): 1717–1721. Zhang J Q, Liu Y Y, He Y, et al. Effect of SO2 on the catalytic performance of CoH-ZSM-5 for selective catalytic reduction of NO by CH4[J]. Environmental Science, 2006, 27(9): 1717–1721. |
| [11] | Lónyi F, Solt H E, Pászti Z, et al. Mechanism of NO-SCR by methane over Co, H-ZSM-5 and Co, H-mordenite catalysts[J]. Applied Catalysis B:Environmental, 2014, 150-151: 218–229. DOI: 10.1016/j.apcatb.2013.12.024 |
| [12] | Skarlis S A, Berthout D, Nicolle A, et al. Combined IR spectroscopy and kinetic modeling of NOx storage and NO oxidation on Fe-BEA SCR catalysts[J]. Applied Catalysis B:Environmental, 2014, 148-149: 446–465. DOI: 10.1016/j.apcatb.2013.11.018 |
| [13] | Lukyanov D B, Sill G, Ditri J L, et al. Comparison of catalyzed and homogeneous reactions of hydrocarbons for selective catalytic reduction(SCR) of NOx[J]. Journal of Catalysis, 1995, 153(2): 265–274. DOI: 10.1006/jcat.1995.1129 |
| [14] | Ren L L, Zhang T, Liang D B, et al. Effect of addition of Zn on the catalytic activity of a Co/HZSM-5 catalyst for the SCR of NOx with CH4[J]. Applied Catalysis B:Environmental, 2002, 35(4): 317–321. DOI: 10.1016/S0926-3373(01)00261-2 |
| [15] | Pan H, Guo Y H, Bi H T. NOx adsorption and reduction with C3H6 over Fe/zeolite catalysts:effect of catalyst support[J]. Chemical Engineering Journal, 2015, 280: 66–73. DOI: 10.1016/j.cej.2015.05.093 |
| [16] | Delahay G, Valade D, Guzmán-Vargas A, et al. Selective catalytic reduction of nitric oxide with ammonia on Fe-ZSM-5 catalysts prepared by different methods[J]. Applied Catalysis B:Environmental, 2005, 55(2): 149–155. DOI: 10.1016/j.apcatb.2004.07.009 |
| [17] | Stranick M A, Houalla M, Hercules D M. Spectroscopic characterization of TiO2/Al2O3 and CoAl2O3-TiO2 catalysts[J]. Journal of Catalysis, 1987, 106(2): 362–368. DOI: 10.1016/0021-9517(87)90247-8 |
| [18] | Zsoldos Z, Guczi L. Structure and catalytic activity of alumina supported platinum-cobalt bimetallic catalysts. 3. Effect of treatment on the interface layer[J]. The Journal of Physical Chemistry, 1992, 96(23): 9393–9400. DOI: 10.1021/j100202a061 |
| [19] | Boroń P, Chmielarz L, Gurgul J, et al. BEA zeolite modified with iron as effective catalyst for N2O decomposition and selective reduction of NO with ammonia[J]. Applied Catalysis B:Environmental, 2013, 138-139: 434–445. DOI: 10.1016/j.apcatb.2013.03.022 |
| [20] | Reddy G K, Boolchand P, Smirniotis P G. Unexpected behavior of copper in modified ferrites during high temperature WGS reaction-Aspects of Fe3+ ↔ Fe2+ redox chemistry from M ssbauer and XPS studies[J]. The Journal of Physical Chemistry C, 2012, 116(20): 11019–11031. DOI: 10.1021/jp301090d |
| [21] | Carrillo A I, Serrano E, Luque R, et al. Microwave-assisted catalysis by iron oxide nanoparticles on MCM-41:effect of the support morphology[J]. Applied Catalysis A:General, 2013, 453: 383–390. DOI: 10.1016/j.apcata.2012.12.041 |
| [22] | 陈艳平, 程党国, 陈丰秋, 等. 非Cu基金属负载分子筛上碳氢化合物选择性催化还原氮氧化物[J]. 化学进展, 2013, 25(12): 2011–2019. Chen Y P, Cheng D G, Chen F Q, et al. Selective catalytic reduction of NOx by hydrocarbons over copper-free metal supported zeolite[J]. Progress in Chemistry, 2013, 25(12): 2011–2019. |
| [23] | Lobree L J, Hwang I C, Reimer J A, et al. An in situ infrared study of NO reduction by C3H8 over Fe-ZSM-5[J]. Catalysis Letters, 1999, 63(3-4): 233–240. |
| [24] | Zhao Y F, Zhao B, Zhuo Y Q, et al. A study of the mechanism of iron-based sulfate catalyst for selective catalytic reduction of NO with NH3[J]. Asia-Pacific Journal of Chemical Engineering, 2012, 7(4): 581–589. DOI: 10.1002/apj.v7.4 |
| [25] | Zhang H Y, Li N, Li L, et al. Selective catalytic reduction of NO with CH4 over In-Fe/sulfated zirconia catalysts[J]. Catalysis Letters, 2011, 141(10): 1491–1497. DOI: 10.1007/s10562-011-0681-4 |
| [26] | Li L D, Guan N J. HC-SCR reaction pathways on ion exchanged ZSM-5 catalysts[J]. Microporous and Mesoporous Materials, 2009, 117(1-2): 450–457. DOI: 10.1016/j.micromeso.2008.07.021 |
| [27] | Jing G H, Li J H, Yang D, et al. Promotional mechanism of tungstation on selective catalytic reduction of NOx by methane over In/WO3/ZrO2[J]. Applied Catalysis B:Environmental, 2009, 91(1-2): 123–134. DOI: 10.1016/j.apcatb.2009.05.015 |
| [28] | Lónyi F, Solt H E, Valyon J, et al. An operando DRIFTS study of the active sites and the active intermediates of the NO-SCR reaction by methane over In, H-and In, Pd, H-zeolite catalysts[J]. Applied Catalysis B:Environmental, 2010, 100(1-2): 133–142. DOI: 10.1016/j.apcatb.2010.07.023 |
| [29] | Pan H, Jian Y F, Yu Y K, et al. Promotional mechanism of propane on selective catalytic reduction of NOx by methane over In/H-BEA at low temperature[J]. Applied Surface Science, 2016, 390: 608–616. DOI: 10.1016/j.apsusc.2016.08.156 |
| [30] | Pietrzyk P, Dujardin C, Góra-Marek K, et al. Spectroscopic IR, EPR, and operando DRIFT insights into surface reaction pathways of selective reduction of NO by propene over the Co-BEA zeolite[J]. Physical Chemistry Chemical Physics, 2012, 14(7): 2203–2215. DOI: 10.1039/C1CP23038G |
| [31] | Wang B, Wu X D, Ran R, et al. Participation of sulfates in propane oxidation on Pt/SO42-/CeO2-ZrO2 catalyst[J]. Journal of Molecular Catalysis A:Chemical, 2012, 361-362: 98–103. DOI: 10.1016/j.molcata.2012.05.007 |