 2016, Vol. 37
2016, Vol. 37



近年来,利用基于硫酸根自由基(SO4 ·-)的高级氧化技术处理水中的难降解有机污染物已成为国内外高级氧化研究的前沿和热点[1, 2, 3]. SO4 ·-具有较高的氧化还原电位(E0为2.6-3.2 V)[4, 5, 6],可通过加成、 取代、 电子转移等方式和有机物分子反应,使之降解成为低毒或无毒的小分子,甚至矿化成为 CO2、 H2O 等无机物质[7, 8, 9, 10]. SO4 ·-的反应活性受pH的限制小,应用范围广. SO4 ·-的产生主要有两种方式:一种是物理方法,主要由过一硫酸盐(peroxymonosulfate,PMS)或过二硫酸盐(peroxydisulfate,PDS)通过辐射或高温分解得到[11, 12, 13]; 另外一种是化学方法,一般用过渡金属离子催化分解过一硫酸盐得到[7, 14, 15, 16]. 采用过渡金属活化PMS产生SO4 ·-在常温常压下即可进行,不需要消耗额外的能量,相比热和紫外光等活化方式具有更大的优势,因而得到广泛的应用. 虽然目前已发现多种过渡金属对PMS具有活化作用,但是Co2+仍被认为是活化性能最好的过渡金属离子[17, 18, 19].
自然环境中Br-和Cl-等卤素离子广泛存在,特别是沿海地区由于海水入侵和地面沉降,地下水和地表水中Br-、 Cl-的存在更为普遍. 因此,具有强氧化性的SO4 ·-在降解污染物的同时,也会与这些无机卤素离子发生反应. 以Br-为例,SO4 ·-可以将其氧化成Br ·(E0为1.62 V),Br ·可以与Br-反应生成Br2 ·-,Br ·、 Br2 ·-相互耦合可生成Br2[20, 21, 22]. 这些反应产生的Br ·、 Br2 ·-及自由溴(包括Br2和HBrO)化学性质活泼,理论上可以与水或土壤中的天然有机质(NOM)反应生成溴代有机物. 这些溴代有机物的产生过程和氯化消毒过程中产生的卤代消毒副产物(disinfection by-products,DBPs)相似. DBPs是对饮用水消毒时,消毒剂(氯气)与水中NOM发生反应生成的卤代副产物. 大多数DBPs具有致癌、 致畸、 致突变的“三致”毒性[23]. 目前被鉴定的DBPs有600多种,其中生成量最大的为三卤甲烷(trihalomethanes,THMs)和卤代乙酸(haloaceficacids,HAAs)[24, 25, 26]. 鉴于DBPs对人类健康具有危害,很多国家对饮用水中的DBPs提出了控制标准. 美国环保署规定饮用水中THMs和HAAs的最大允许量(MCL)分别为60 μg ·L-1和80 μg ·L-1. 我国《生活饮用水卫生标准》规定氯仿、 一溴二氯甲烷、 二溴一氯甲烷、 三溴甲烷限值分别为60、 60、 100、 100 μg ·L-1; HAAs中氯乙酸和三氯乙酸限值分别为50 μg ·L-1和100 μg ·L-1[27].
根据热力学分析,在过硫酸盐高级氧化过程中,会产生活泼的卤自由基和自由卤,它们可以和环境中的NOM反应,从而导致卤仿、 卤乙酸等卤代DBPs的生成. 本课题组在先前的工作中已证实了热活化PDS氧化过程中DBPs的生成[11]. Wang等[28]也报道了Br-存在条件下,CuFe2O4活化PMS过程中有溴代消毒副产物(Br-DBPs)生成. 然而,在基于SO4 ·-的高级氧化过程中,卤素是如何转化并加到有机物分子上,鲜见报道. 由于天然腐殖质结构的不确定性,在DBPs生成的研究中常用苯酚类物质作为模型化合物来模拟NOM的结构[29]. 本文选用苯酚作为NOM的模型化合物,深入研究在Co2+/PMS氧化过程中DBPs生成的途径和机制,以期为全面评价过硫酸盐高级氧化工艺在污染控制方面的应用可行性提供依据.
1 材料与方法 1.1 材料与试剂使用试剂均为分析纯及以上纯度. THMs混标、 HAAs混标、 1,2,3-三氯丙烷、 1,2-二溴丙烷、 2-溴苯酚、 4-溴苯酚、 2,4,6-三溴苯酚购自Sigma-Aldrich公司. 甲基叔丁基醚(MTBE)购自Fisher公司,色谱纯. N,N-二乙基对苯二胺盐酸盐(DPD)、 苯酚、 过硫酸氢钾(KHSO5)、 溴化钾(KBr)、 硫酸钴(CoSO4)、 氯化钠(NaCl)、 亚硫酸钠(Na2SO3)购自阿拉丁公司. 配制试剂等所有实验用水均为Milli-Q超纯水(Millipore,18.2 MΩ ·cm).
1.2 实验设计 1.2.1 DBPs的生成所有反应均在45 mL EPA瓶中、 室温下进行. 反应溶液中加入10 mmol ·L-1磷酸盐缓冲液调节pH为6.0. 反应开始前,先在EPA瓶中加入浓度分别为0.05 mmol ·L-1、 0.2 mmol ·L-1的苯酚和Br-,然后加入一定量的PMS和Co2+,设置PMS浓度为1 mmol ·L-1、 5 mmol ·L-1,Co2+浓度为0、 5 μmol ·L-1,拧紧瓶盖,开始计时. 反应过程中保证溶液充满EPA瓶,待反应分别进行至2、 4、 6、 8、 10、 12、 24、 48 h时,向瓶中加入过量的Na2SO3终止反应,样品放冰箱待进一步处理. 每个反应时间点的样品准备两份,分别用于检测THMs、 HAAs.
THMs分离富集和分析参照US EPA 551.1标准方法进行. 样品经MTBE萃取后用GC-ECD检测. 检测条件如下:进样口温度为200℃,检测器温度为290℃,载气为99.999%高纯氮气,流速为27 cm ·s-1. 初始柱温为35℃,保持11 min; 10 ℃ ·min-1升至120℃,保持8 min; 10 ℃ ·min-1升至150℃,保持5 min,共计35.5 min. HAAs具有非挥发性,参照US EPA 552.2标准方法,样品经MTBE萃取后用酸化甲醇作衍生剂将HAAs酯化后用GC-ECD进行分析. 检测条件:进样口温度为200℃,检测器温度为260℃,载气为99.999%高纯氮气,流速为27 cm ·s-1. 初始柱温为35℃,保持10 min; 先以5 ℃ ·min-1的速度升至75℃,保持15 min; 再以5 ℃ ·min-1的速度升至100℃,保持5 min,最后以5 ℃ ·min-1的速度升至135℃,保持2 min,共计52 min.
1.2.2 氯离子对DBPs生成的影响反应溶液中同时加入Br-和Cl-,固定其总量为0.2 mmol ·L-1,调节Br-/Cl-分别为1 ∶0、 8 ∶2、 6 ∶4、 4 ∶6、 2 ∶8、 0 ∶1,其余反应条件同上. 反应8 h,检测DBPs的生成.
1.2.3 pH对DBPs生成的影响固定反应溶液中PMS浓度为5 mmol ·L-1,Co2+浓度为0、 5 μmol ·L-1,通过NaOH和H2SO4调节反应液pH值分别为2.6、 3.4、 5.4、 7.4、 8.5、 9.6,其余反应条件同上,检测DBPs的生成.
1.2.4 中间产物的测定分别在加Co2+和不加Co2+条件下,保持反应液中含有0.05 mmol ·L-1 苯酚,0.2 mmol ·L-1 Br-,1 mmol ·L-1 PMS,在EPA瓶中启动反应. 到设定反应时间后,样品用Agilent 6410B三重四级杆质谱检测反应过程中产生的中间产物. 电喷雾离子源(ESI)设为负离子模式,氮气(≥ 99.995%)作干燥气,流速10 mL ·min-1,干燥气温度为350℃,毛细管电压为4.0 kV,碎裂电压设为125 V,喷雾器压力为0.276 MP(40 psi).
1.2.5 自由溴的生成自由溴参照国标 GB 11898-89《水质游离氯和总氯的测定》中规定的N,N-二乙基对苯二胺(DPD)分光光度法进行测定. 在EPA瓶中启动反应开始计时,反应条件同上,每隔5 min取1 mL反应液,稀释5倍,迅速加入1 mL pH为6.5的缓冲液和1 mL DPD,用Varian Cary50 紫外-可见分光光度计检测其在510 nm处的吸光度.
2 结果与分析 2.1 自由溴的生成有报道指出,SO4 ·-可以将Br-氧化为Br2和HBrO等活性物质. 从热力学角度分析,SO4 ·-的氧化还原电位高达2.5-3.1 V,而Br ·的氧化还原电位为1.62 V[30]. 当Br-与 SO4 ·-作用会迅速生成具有反应活性的Br ·,然后进一步生成Br2 ·-、 Br2和HBrO等活性物质. 本研究Co2+活化PMS反应溶液中自由溴的生成如图 1所示. 自由溴生成量随着反应时间的延长呈现先增加后减少的趋势. 反应时间为0.5 h时生成量达到最大,为82 μmol ·L-1,之后随反应时间的延长而逐渐减少,24 h时降至10 μmol ·L-1. 这一变化规律表明,在Co2+活化PMS过程中,自由溴仅仅是Br-转化过程中的中间产物,而不是最终产物. 在UV活化PMS过程中类似的Br-转化途径已经有相关报道,在此过程中Br-最终被氧化为BrO3-[21].
 | 反应条件:苯酚=0.05 mmol ·L-1; Br-=0.2 mmol ·L-1; PMS=1 mmol ·L-1; Co2+=0、 5 μmol ·L-1; t=24 h图 1 Co2+/PMS氧化过程中自由溴的生成 Fig. 1 Formation of free bromine during the oxidation process in the Co2+/PMS system |
由图 1可知,加入苯酚后,自由溴也出现了类似的生成规律,但是浓度明显降低. 这是由于苯酚通过以下两种途径参与到了复杂的反应中. 第一,生成的自由溴直接与苯酚反应而被消耗. Gallard等[31]对6种苯酚类物质与自由溴的反应速率进行了比较,结果表明在中性条件下,苯酚溴化的二级反应速率常数为1.0×106 M-1 ·s-1[32],说明二者极易反应; 第二,部分活化产生的SO4 ·-和苯酚反应被消耗,导致用于氧化Br-的SO4 ·-减少,自由溴的生成受到抑制. 综上所述,苯酚的加入既会消耗产生的自由溴,同时也会通过和SO4 ·-反应进而抑制自由溴的生成.
值得注意的是,反应液中不含Co2+时,同样检测到了自由溴的生成. 自由溴的生成量随着反应时间的延长而增加,在3 h时达到最大. 这一结果说明了PMS本身可以将Br-氧化成自由溴. PMS是一种强氧化剂(HSO5-/HSO4-,1.82 V),其氧化还原电位高于HBrO(Br-/HBrO,1.338 V)[33]. 但对比Co2+/PMS反应过程,不含Co2+的溶液中Br-的转化速率较为缓慢. 将数据用假一级反应动力学模型进行拟合,可得到PMS转化Br-的二级反应速率常数为0.17 M-1 ·s-1,远低于SO4 ·-与Br-的反应速率常数(3.5×109 M-1 ·s-1)[34]. 与Co2+/PMS 氧化过程相比,PMS本身的氧化能力有限,自由溴不能被进一步氧化,因此不含Co2+的反应液中自由溴的浓度随反应的进行并没有明显降低. 同时苯酚的加入明显降低了反应液中自由溴的生成量,主要是由于自由溴与苯酚反应而被消耗.
2.2 溴代中间产物的鉴定SO4 ·-和活性溴物种对苯酚的双重作用,势必会导致苯酚的转化,尤其是发生溴化反应,生成溴代产物. 本研究采用MS对反应过程中的中间产物进行了检测,结果如图 2所示. 首先根据MS图谱中溴元素的特征同位素指纹来判读是否生成了溴代产物,在此基础上通过分子量推测产物的可能结构,然后通过和标样比对进行确证. 由于Br-具有两个天然同位素(79 Br 和81 Br),丰度比接近于1 ∶1,因此含溴有机物分子的同位素峰簇的相对丰度符合二项式 (1+1)n的展开,其中n指分子中所含溴原子的个数. 如图 2(b)所示,Co2+活化PMS反应溶液中出现了比例分别为1 ∶1、 1 ∶2 ∶1、 1 ∶3 ∶3 ∶1的m/z 171/173、 m/z 265/267/269、 m/z 327/329/331/333特征吸收峰簇,分别符合含有1、 2、 3个Br原子的分子的同位素峰分布规律. 经对比标准样品确定m/z 171/173对应的产物是2-溴苯酚和4-溴苯酚; m/z 327/329/331/333对应的产物为2,4,6-三溴苯酚. 根据分子量、 同位素丰度比例推测,m/z 265/267/269可能对应于二溴对苯二酚或二溴邻苯二酚. 除了上述3种溴代苯酚外,Co2+活化PMS反应液中还出现了比例为1 ∶1的m/z 187/189特征吸收峰簇. 根据其分子量,推断该物质为2-溴对苯二酚或4-溴邻苯二酚. 这几种中间产物的分子结构见表 1. SO4 ·-是亲 电物质[35],与苯酚作用时苯酚分子中的电子先转移到SO4 ·-上或通过加成消除反应,从而生成苯酚阳离子. 苯酚阳离子既可以通过羟基化生成对苯二酚或者邻苯二酚,也可以与HBrO等活性溴物质作用生成溴代中间产物. 生成的这部分对苯二酚或邻苯二酚也可以直接与HBrO等活性溴物质反应生成对应的溴代产物.
 | 反应条件:苯酚=0.05 mmol ·L-1; Br-=0.2 mmol ·L-1; PMS=1 mmol ·L-1; 负离子状态扫描图 2 Br-存在下Co2+/PMS氧化苯酚过程中间产物的质谱图 Fig. 2 MS of phenol treated with PMS in the presence of Br- |
| 表 1 Br-存在下Co2+/PMS氧化苯酚过程中间产物的 荷质比、 同位素丰度比例及其分子结构 Table 1 Molecular structures,m/z values and isotopic abundance ratios of the possible intermediates in Co2+/PMS oxidation of phenol in the presence of Br- |
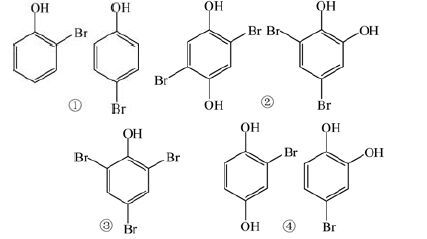
反应液中不加入Co2+时,仅检测到了3种溴代产物,分别是m/z 171/173、 m/z 265/267/269、 m/z 327/329/331/333,它们对应于一溴苯酚、 二溴苯二酚、 三溴苯酚,这是苯酚和Br2/HBrO反应的结果.
2.3 Br-DBPs的生成上述过程中产生的溴代苯酚在溶液中不会稳定存在,在SO4 ·-或活性溴物种的作用下,会发生开环断裂,形成更小分子量的溴代产物. 这些反应过程和氯化消毒过程中产生的卤代反应过程类似. 因此,最终产物很可能是卤仿和卤乙酸等小分子物质. 本研究检测到的产物主要是三溴甲烷(CHBr3)和二溴乙酸(DBAA),它们的生成规律如图 3所示. 加Co2+和不加Co2+条件下,Br-DBPs生成量随时间的变化趋势呈现明显的差异. 加Co2+条件下,生成Br-DBPs的速率较快,随时间的延长呈现先增加后减少的趋势,在8 h时达到最大值. 而不加Co2+条件下,Br-DBPs(尤其是DBAA)的生成呈现明显的滞后,其生成量随时间的延长而增加,反应时间8 h达到最大,12 h后保持基本稳定. 以三溴甲烷为例,PMS为1 mmol ·L-1条件下,反应液中加入Co2+时,在8 h即达到最大值10.3 μmol ·L-1,12 h时降低至7.2 μmol ·L-1,之后的反应时间中变化不大. 不含Co2+时,生成量随着时间的增加而增加,10 h时达到最大值12.2 μmol ·L-1,并维持在此水平上. 二溴乙酸呈现出相同的变化趋势. 加Co2+条件下,其生成量6 h内呈现逐渐增加的趋势,在6 h达到最大值14.6 μmol ·L-1. 随着时间的延长逐渐降低,12 h降低至10 μmol ·L-1. 随着时间的进一步延长,其降低的趋势变得缓慢. 不加Co2+条件下,反应时间为8 h时达到最大值15.4 μmol ·L-1,随后的时间中保持基本稳定. 加Co2+和不加Co2+条件下DBPs生成情况的对比可以看出,SO4 ·-会将生成的DBPs进一步氧化.
 | 反应条件:苯酚=0.05 mmol ·L-1; Br=0.2 mmol ·L-1; t=48 h图 3 Br-存在下Co2+/PMS氧化苯酚过程中DBPs的生成 Fig. 3 Formation of DBPs in the Co(Ⅱ)/PMS system during phenol oxidation in the presence of Br- |
不含Co2+的溶液中Br-的转化主要由PMS推动,而含有Co2+的溶液中推动反应的主要是SO4 ·-,SO4 ·-的氧化还原电位远高于PMS,可以降解多种卤代有机物. 当SO4 ·-足量的情况下,生成的DBPs会进一步又被SO4 ·-分解. 因此在Co2+/PMS反应液中三溴甲烷和二溴乙酸的含量随着反应的进行先增加后减少,48 h时三溴甲烷和二溴乙酸的含量分别为5.7 μmol ·L-1和6.1 μmol ·L-1. 当PMS为5 mmol ·L-1时,DBPs的降解更加明显,反应48 h溶液中三溴甲烷和二溴乙酸的量仅为0.1 μmol ·L-1和1.4 μmol ·L-1.
2.4 氯离子对DBPs生成的影响Cl-是水中普遍存在的无机离子,由于Cl-与Br-的性质极其相似,因此反应溶液中含有Cl-势必会对Br-DBPs的生成造成影响. 本研究向反应溶液中同时加入Br-和Cl-,固定其总量为0.2 mmol ·L-1,调节Br-/Cl-分别为1 ∶0、 8 ∶2、 6 ∶4、 4 ∶6、 2 ∶8、 0 ∶1,反应8 h,DBPs的生成如图 4所示. Cl-的加入明显影响了DBPs的种类和产量,但是仍以Br-DBPs为主要产物; 反应过程中DBPs总生成量随Cl-/Br-比的增大而减小,但含氯DBPs的生成量随着Cl-浓度的增加而增加; 而当反应液中只含Cl-时,没检测到任何DBPs. 不论Br-/Cl-比例如何,反应过程中生成的THMs仍以溴仿为主,同时有少量的一氯二溴甲烷(<0.26 μmol ·L-1)和二氯一溴甲烷(<0.25 μmol ·L-1),以及微量的氯仿(<0.01 μmol ·L-1)生成. HAAs的生成规律和THMs相似,反应产物仍以二溴乙酸(DBAA)为主,伴随着Cl-浓度的增加,HAAs的种类也在增加,包括一溴乙酸(MBAA)、 二溴乙酸(DBAA)、 二氯一溴乙酸(DCBAA)、 一氯二溴乙酸(CDBAA)、 二氯乙酸(DCAA)、 一氯一溴乙酸(CBAA)均有生成. 如图 4所示,当PMS和Co2+初始浓度分别为5 mmol ·L-1和5 μmol ·L-1时,随着Cl-浓度从0增加至0.2 mmol ·L-1,反应液中DBPs总生成量迅速降低,其中THMs由8.7 μmol ·L-1降至0.01 μmol ·L-1,HAAs的生成量由21.2 μmol ·L-1降至2.6 μmol ·L-1.
 | 反应条件:苯酚=0.05 mmol ·L-1; PMS=5 mmol ·L-1; Co2+=5 μmol ·L-1; Cl-=0-0.2 mmol ·L-1; t=8 h图 4 DBPs的生成量随Cl-浓度的变化 Fig. 4 Variation of DBPs formation with different Cl- concentration |
综上所述,Co2+/PMS反应过程中同时含有Br-和Cl-的情况下,有机溴的产量远高于有机氯,这一方面跟Br-和Cl-被SO4 ·-氧化的难易程度有关,同时还体现了自由溴和自由氯对苯酚的反应活性的不同. 苯酚的卤化是一个亲电取代反应,自由溴的亲电性强于自由氯,因此苯酚溴化的速率远高于氯化[36]. 因此在竞争苯酚过程中,自由氯相对弱势,导致生成更多的溴代消毒副产物和微量的氯代消毒副产物.
2.5 pH对DBPs生成的影响pH对过硫酸盐氧化过程中Br-DBPs生成的影响如图 5所示. pH越高,溴仿和二溴乙酸的生成量越低. 当pH为2.6时,溴仿和二溴乙酸的生成量最大,分别为2.4μmol ·L-1和3.2 μmol ·L-1. pH为9.6时,溶液中溴仿和二溴乙酸生成量分别降低至0.78μmol ·L-1和0.46 μmol ·L-1以下,不及pH为2.6时的1/3. 这是由于酸性条件下更容易生成SO4 ·-,使Br-迅速转化为自由溴,进而产生更多DBPs. 同时在碱性条件下,SO4 ·-与OH-快速反应生成OH ·. OH ·在水溶液中的半衰期较短,容易被快速淬灭,不能有效地将溶液中的Br-转化. 因此在碱性条件下Br-DBPs的生成量明显降低.
 | 反应条件:苯酚=0.05 mmol ·L-1; Br-=0.2 mmol ·L-1; PMS=5 mmol ·L-1; Co2+=0、 5 μmol ·L-1; t=8、 24 h图 5 DBPs的生成量随pH的变化 Fig. 5 pH-dependent formation of DBPs in the Co(Ⅱ)/PMS system |
Co2+活化PMS过程中,Br-在SO4 ·-作用下转化为Br ·、 Br2 ·-、 Br2及HOBr等活性溴. 这些活性溴会引起苯酚的溴化,导致溴代酚以及Br-DBPs(主要包括三溴甲烷和二溴乙酸)的生成. 在不加Co2+的情况下Br-DBPs也有生成,并且生成量更高. 反应时间、 钴离子浓度和pH等反应条件均对Br-DBPs的生成速率、 生成量以及分布有明显影响. Cl-的加入明显影响DBPs的种类和生成量,但是仍以Br-DBPs为主要产物. 反应过程中DBPs总生成量随Cl-/Br-比的增大而减小,且含氯原子的消毒副产物生成量随着Cl-浓度的增加而增加. 由于Cl-和Br-在环境介质中广泛存在,在利用产生SO4 ·-的高级氧化工艺降解污染物的同时,存在产生卤代消毒副产物的可能,这一问题应引起关注.
| [1] | Bandala E R, Peláez M A, Salgado M J, et al. Degradation of sodium dodecyl sulphate in water using solar driven Fenton-like advanced oxidation processes[J]. Journal of Hazardous Materials, 2008, 151 (2-3): 578-584. |
| [2] | Ikehataa K, El-Din M G. Degradation of recalcitrant surfactants in wastewater by ozonation and advanced oxidation processes: a review[J]. Ozone Science & Engineering, 2004, 26 (4): 327-343. |
| [3] | 龙安华,雷洋,张晖. 活化过硫酸盐原位化学氧化修复有机污染土壤和地下水[J]. 化学进展, 2014, 26 (5): 898-908. |
| [4] | Fang J Y, Shang C. Bromate formation from bromide oxidation by the UV/Persulfate process[J]. Environmental Science & Technology, 2012, 46 (16): 8976-8983. |
| [5] | Furman O S, Teel A L, Watts R J. Mechanism of base activation of persulfate[J]. Environmental Science & Technology, 2010, 44 (16): 6423-6428. |
| [6] | Liang C J, Lee I L, Hsu I Y, et al. Persulfate oxidation of trichloroethylene with and without iron activation in porous media[J]. Chemosphere, 2008, 70 (3): 426-435. |
| [7] | Anipsitakis G P, Dionysiou D D. Degradation of organic contaminants in water with sulfate radicals generated by the conjunction of peroxymonosulfate with cobalt[J]. Environmental Science & Technology, 2003, 37 (20): 4790-4797. |
| [8] | Xu L, Yuan R X, Guo Y G, et al. Sulfate radical-induced degradation of 2, 4, 6-trichlorophenol: A de novo formation of chlorinated compounds[J]. Chemical Engineering Journal, 2013, 217: 169-173. |
| [9] | 欧阳磊, 丁耀彬, 朱丽华, 等. 钴掺杂铁酸铋活化过硫酸盐降解水中四溴双酚A的研究[J]. 环境科学, 2013, 34 (9): 3507-3512. |
| [10] | 张轶, 黄若男, 王晓敏, 等. TiO2光催化联合技术降解苯酚机制及动力学[J]. 环境科学, 2013, 34 (2): 596-603. |
| [11] | Liang C J, Bruell C J. Thermally activated persulfate oxidation of trichloroethylene: Experimental investigation of reaction orders[J]. Industrial & Engineering Chemistry Research, 2008, 47 (9): 2912-2918. |
| [12] | Lu J H, Wu J W, Ji Y F, et al. Transformation of bromide in thermo activated persulfate oxidation processes[J]. Water Research, 2015, 78: 1-8. |
| [13] | Zhong J B, Di M, Zhao H, et al. Photocatalytic decolorization of methyl orange solution with potassium peroxydisulfate[J]. Central European Journal of Chemistry, 2008, 6 (2): 245-252. |
| [14] | 李欢旋, 万金泉, 马邕文, 等. 不同粒径零价铁活化过硫酸钠氧化降解酸性橙7的影响及动力学研究[J]. 环境科学, 2014, 35 (9): 3422-3429. |
| [15] | Gilbert B C, Stell J K, Jeff M. Electron spin resonance studies of the effect of copper(Ⅱ) and copper(Ⅰ) on the generation and reactions of organic radicals formed from the fenton reaction and the TiⅢ-H2O2 and TiⅢ-S2O82- redox couples[J]. Journal of the Chemical Society, Perkin Transactions 2, 1988, 10 (10): 1867-1873. |
| [16] | Anipsitakis G P, Dionysiou D D. Radical generation by the interaction of transition metals with common oxidants[J]. Environmental Science & Technology, 2004, 38 (13): 3705-3712. |
| [17] | Chan K H, Chu W. Degradation of atrazine by cobalt-mediated activation of peroxymonosulfate: different cobalt counteranions in homogenous process and cobalt oxide catalysts in photolytic heterogeneous process[J]. Water Research, 2009, 43 (9): 2513-2521. |
| [18] | Rivas F J, Gimeno O, Borallho T. Aqueous pharmaceutical compounds removal by potassium monopersulfate. Uncatalyzed and catalyzed semicontinuous experiments[J]. Chemical Engineering Journal, 2012, 192: 326-333. |
| [19] | Yang Q J, Choi H, Chen Y J, et al. Heterogeneous activation of peroxymonosulfate by supported cobalt catalysts for the degradation of 2, 4-dichlorophenol in water: the effect of support, cobalt precursor, and UV radiation[J]. Applied Catalysis B: Environmental, 2008, 77 (3-4): 300-307. |
| [20] | Fang G D, Dionysiou D D, Wang Y, et al. Sulfate radical-based degradation of polychlorinated biphenyls: effects of chloride ion and reaction kinetics[J]. Journal of Hazardous Materials, 2012, 227-228: 394-401. |
| [21] | Lutze H V, Bakkour R, Kerlin N, et al. Formation of bromate in sulfate radical based oxidation: mechanistic aspects and suppression by dissolved organic matter[J]. Water Research, 2014, 53: 370-377. |
| [22] | Acero J L, Piriou P, Von Gunten U. Kinetics and mechanisms of formation of bromophenols during drinking water chlorination: assessment of taste and odor development.[J]. Water Research, 2005, 39 (13): 2979-2993. |
| [23] | Richardson S D, Plewa M J, Wagner E D, et al. Occurrence, genotoxicity, and carcinogenicity of regulated and emerging disinfection by-products in drinking water: a review and roadmap for research[J]. Mutation Research/Reviews in Mutation Research, 2007, 636 (1-3): 178-242. |
| [24] | Karanfil T, Krasner S W, Westerhoff P, et al. Disinfection by-products in drinking water: occurrence, formation, health effects, and control (ACS symposium series) [M]. Washington: American Chemical Society, 2008. 995. |
| [25] | World Health Organization. Guidelines for drinking-water quality[R]. Geneva: World Health Organization, 1996. |
| [26] | 张岚, 陈昌杰, 陈亚妍. 我国生活饮用水卫生标准[J]. 中国公共卫生, 2007, 23 (11): 1281-1282. |
| [27] | 张小璐, 杨宏伟, 王小, 等. 消毒副产物生成的温度影响和动力学模型[J]. 环境科学, 2012, 33 (11): 4046-4051. |
| [28] | Wang Y R, Roux J L, Zhang T, et al. Formation of brominated disinfection byproducts from natural organic matter isolates and model compounds in a sulfate radical-based oxidation process[J]. Environmental Science & Technology, 2014, 48 (24): 14534-14542. |
| [29] | 王小文, 张晓健, 陈超, 等. 芳香类有机物生成氯化消毒副产物特性及其与化学结构的关系[J]. 环境科学, 2006, 27 (8): 1603-1607. |
| [30] | Huie R E, Clifton C L, Neta P. Electron transfer reaction rates and equilibria of the carbonate and sulfate radical anions[J]. International Journal of Radiation Applications and Instrumentation. Part C. Radiation Physics and Chemistry, 1991, 38 (5): 477-481. |
| [31] | Gallard H, Von Gunten U. Chlorination of phenols: kinetics and formation of chloroform[J]. Environmental Science & Technology, 2002, 36 (5): 884-890. |
| [32] | Guo G M, Lin F K. The bromination kinetics of phenolic compounds in aqueous solution[J]. Journal of Hazardous Materials, 2009, 170 (2-3): 645-651. |
| [33] | Rastogi A, Al-Abed S R, Dionysiou D D. Sulfate radical-based ferrous-peroxymonosulfate oxidative system for PCBs degradation in aqueous and sediment systems[J]. Applied Catalysis B: Environmental, 2009, 85 (3-4): 171-179. |
| [34] | Buxton G V, Greenstock C L, Helman W P, et al. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (·OH/·O- in aqueous solution[J]. Journal of Physical and Chemical Reference Data, 1988, 17 (2): 513-886. |
| [35] | Gilbert B C, Smith J R L, Taylor P, et al. The interplay of electronic, steric and stereoelectronic effects in hydrogen-atom abstraction reactions of SO4-·, revealed by EPR spectroscopy[J]. Journal of the Chemical Society, Perkin Transactions 2, 1999, (8): 1631-1638. |
| [36] | Lu J H, Benjamin M M, Korshin G V, et al. Reactions of the flavonoid hesperetin with chlorine: a spectroscopic study of the reaction pathways[J]. Environmental Science & Technology, 2004, 38 (17): 4603-4611. |