 2016, Vol. 37
2016, Vol. 37



我国金属硫化矿物资源丰富、 分布广泛,硫化矿物氧化引发土壤酸化,导致土壤肥力下降、 有毒金属离子淋溶而污染环境[1]. 硫化矿物在酸性环境中溶解度增强易形成水溶性硫化物; 石油、 化工、 造纸和印染等排放的废水中多有高浓度水溶性硫化物. 硫化物具有强的腐蚀性和毒性,影响材料结构与性能、 威胁人体健康和环境微生物生长与繁殖[2, 3]. 因此,水溶性硫化物的氧化行为、 迁移转化与归趋一直是环境科学与土壤科学关注的热点.
在土壤中广泛存在的氧化锰矿物影响污染物的环境化学行为,如吸附氧化有毒金属离子(如Pb2+、 Cu2+和AsO2-等)、 有机化合物(如苯酚、 抗生素和农药)[3, 4, 5, 6, 7, 8]. 锰氧化物与水溶性硫化物的氧化还原行为也得到了广泛研究. 氧化锰矿物晶体结构类型和反应条件不同,其产物也不尽相同[9, 10, 11, 12, 13, 14, 15]. 晶体结构类型影响氧化锰氧化水溶性硫化物的速率[9, 10, 11, 12, 13, 14, 15],而Mn(Ⅲ)含量影响锰氧化物的氧化活性[15],进一步研究表明其氧化活性还受氧化锰晶体内Mn(Ⅲ)位点影响,如酸性水钠锰矿中Mn(Ⅲ)位于八面体空位的上下方,即层间Mn(Ⅲ)与水溶性硫化物接触反应几率增加,即使其含量低也表现出高的氧化活性[10]. 现有研究多关注氧化活性强的锰氧化物(如水钠锰矿、 锰钾矿和钙锰矿)与硫化物的氧化还原行为,而对氧化活性弱、 低价态的水锰矿关注较少[9, 10, 11, 12, 13, 14, 15]. 水锰矿(γ-MnOOH)能稳定存在于土壤与沉积物中,氧化活性高的氧化锰矿物还原可直接生成水锰矿,Mn(OH)2沉淀在有氧环境中可氧化生成亚稳态六方水锰矿(β-MnOOH),其可进一步转化生成水锰矿[10, 16, 17]. 较高氧化活性的锰氧化物,水锰矿参与水溶性硫化物的氧化反应中,反应产物组分是否相同还不明确; 稳定的水锰矿在有氧环境中会否表现出一定的催化活性仍有待研究.
水锰矿参与金属离子和有机污染物的吸附氧化过程,如能有效吸附As、 Zn、 Cr等有毒金属离子[18, 19, 20]. 锰氧化物与水溶性硫化物反应过程也多为关注缺氧体系,而在有氧环境中,水锰矿与水溶性硫化物的反应过程也会有所差异. 为此,本文主要模拟研究了水锰矿与水溶性硫化物的氧化还原行为,考察了初始pH和氧气氛围对反应过程的影响.
1 材料与方法 1.1 水锰矿的制备在碱性条件下采用H2O2氧化MnSO4法合成水锰矿[18, 19]. 将250 mL 1.4 mol ·L-1 MnSO4 ·H2O和250 mL 0.4 mol ·L-1 NH3 ·H2O置于烧杯中磁力搅拌均匀,用恒流泵以3 mL ·min-1逐滴加入90 mL H2O2(30%)溶液,滴加完毕后将烧杯置于105℃的油浴锅中恒温回流反应24 h. 自然冷却后用去离子水洗涤产物至电导率低于15 μS ·cm-1,产物在40℃烘箱内烘干至恒重,研磨过75目筛以备用.
1.2 水锰矿氧化S2-实验在除氧的Na2S溶液(S32-浓度:200 mg ·L-1)中加入0.50 g水锰矿,反应体系(400 mL)中持续通入高纯氮气以维持厌氧环境,在搅拌条件下水浴恒温20℃,反应不同时间段后用注射器抽取3.0 mL的悬浊液,用0.22 μm微孔滤膜过滤,滤液保留待测. 用XRD、 FTIR表征过滤后所得固相产物组分,也即载有湿样品的滤膜紧贴样品池,进行XRD分析; 载有湿样的滤膜经40℃真空烘干后抖落粉末产物进行FTIR表征. 固相产物中单质S经甲醇-水溶液萃取后用高效液相色谱仪分析其含量,滤液中S2-用亚甲基蓝分光光度法测定,S2O2-、 SO32-和SO42-等浓度用离子色谱仪测定. 值得一提的是,氮气流速控制为气泡连续鼓出以保证缺氧环境,单一硫化钠溶液在240 h内S2-损失率不足10%,显著低于其被氧气和水锰矿氧化的转化速率,因此氮气吹脱作用可忽略.
上述体系在反应过程中初始pH值约为12.0,为考察初始阶段pH对反应过程的影响,用稀盐酸和NaOH调控反应体系起始pH分别为10.0和8.0. 为模拟开放有氧环境下其反应过程,在体系中通入氧气维持有氧环境监测中间产物.
1.3 产物组分与理化性质表征所得水锰矿用全自动比表面仪(Quantachrome Autosorb-1)测定比表面积; X-射线衍射仪(Bruker D8 ADVANCE)表征固相产物晶体结构,采用Cu Kα(λ=0.160 54 nm),管压40 kV、 管流40 mA、 步长0.02°,扫速为4(°) ·min3-1; 透射电镜(TEM,Hitachi H-7650)观察样品形貌. 傅里叶变换红外光谱仪(FTIR,Bruker Equinox 55)分析产物的特征官能团. 固相产物中单质硫含量以纯甲醇和含有5%水的甲醇溶液用作洗脱液[12]分离后高效液相色谱仪(Agilent,HPLC-1200)分析测定; 离子色谱(Dionex,ICS-1100)测定S2O2-、 SO32-和SO42-等浓度[10].
2 结果与讨论 2.1 水锰矿氧化S2-
合成氧化锰矿物样品X-射线衍射谱如图 1(a)所示,XRD谱峰与水锰矿标准谱图(γ-MnOOH,JCPDS:41-1379)一致,表明所得产物为纯相水锰矿. 水锰矿为尺寸约200-500 nm的棒状颗粒[图 1(b)],比表面积为42.4 m2 ·g-1.
 | 图 1 水锰矿XRD图谱和透射电镜照片 Fig. 1 XRD patterns and TEM images of synthesized γ-MnOOH |
在S32-浓度为200 mg ·L-1的氮气保护体系中,加入0.5 g水锰矿后初始pH约为12.0,恒温20℃反应不同时间段S2-转化率及氧化产物浓度变化趋势如图 2所示,反应90 min左右,水溶性硫化物(S2-)转化速率达99.7%,也即溶液中S2-基本完全被水锰矿氧化. 与钙锰矿、 水钠锰矿和锰钾矿等高价态锰氧化物相比,水锰矿完全氧化S2-所需时间显著延长,表明其氧化活性较低. 可见,晶体结构类型和化学组分等会影响氧化锰矿物的氧化活性[9, 10]. 单质S为S2-氧化主要产物,反应90 min左右其转化率达85%,在210 min时为92.3%,而后略有下降,可能反应后期进一步被氧化. 液相组分S2O2-、 SO32-和SO432-浓度均较低,其中S2O2-浓度相对较高,且浓度有先增加后降低的趋势,可能是生成的S2O32-分解为单质S和SO32-,而S32-又可与SO2-反应生成单质S[9, 10].
 | 图 2 氮气氛围下0.5 g水锰矿与S2-溶液(400 mL, 200 mg ·L-1)反应产物及S2-的转化率 Fig. 2 Transformation rate from S2- to S2O32-,SO32-, SO42- and the oxidation rate of S2- (200 mg ·L-1,400 mL) |
不同时间段固相产物XRD分析结果如图 3(a)所示,反应20 min后水锰矿还原成Mn(OH)2(JCPDS:12-0696),且反应体系液相中并未检测出Mn2+,可见,初始pH为12.0的强碱环境中新生成的Mn2+易生成Mn(OH)2沉淀. 反应进行60 min后,出现了单质硫α-S8的特征衍射峰(JCPDS:83-2284),且随着反应时间延长,α-S8特征衍射峰增强,表明单质S含量有增加的趋势. 反应进行4-11 h,γ-MnOOH未完全还原消失,除γ-MnOOH、 α-S8和Mn(OH)2的特征衍射峰外,还观察到Mn3O4(JCPDS:16-0350)的特征衍射峰,主要原因是潮湿的Mn(OH)2在空气中被氧化容易转化生成Mn3O4[10].
 | 图 3 氮气氛围下0.5 g水锰矿与S2-溶液(400 mL,200 mg ·L-1)反应不同时间段固相产物XRD图谱和FTIR图谱 Fig. 3 XRD patterns and FITR spectra of solid products from the oxidation rate of S2- (200 mg ·L-1,400 mL) by γ-MnOOH at different time in nitrogen atmosphere |
为进一步阐明水锰矿氧化S2-的反应过程,进一步对固相产物进行FTIR分析,明确其中间产物. 如图 3(b)所示3 431、 1 631 cm-1为H2O的特征吸收峰[21, 22]; 2 083、 1 261、 1 152、 1 085、 1 022、 595、 495、 447 cm-1为γ-MnOOH的吸收振动峰,430 cm-1处为Mn3O4的吸收峰[22, 23]; α-S8的特征吸收峰位于442 cm-1处[24],与γ-MnOOH 447 cm-1峰位重叠,随着反应进行固相产物在该处的吸收峰逐渐宽化说明有单质S生成. 同时观察到γ-MnOOH特征吸收峰逐渐减弱,主要为Mn(OH)2在空气中被氧化成Mn3O4,这与XRD分析结果较为一致.
反应过程中间产物微观形貌如图 4所示. 反应20 min所得固相产物生成部分片状结构产物[图 4(a)],这可能为生成的α-S8[9]. 反应6 h后棒状颗粒物粉化[图 4(b)],由于MnOOH被还原后形成大量形状不规则小颗粒. 结合XRD及TEM分析结果可知,水锰矿经还原多转化为Mn(OH)2.
 | (a)20 min; (b) 360 min图 4 氮气氛围下0.5 g水锰矿与S2-溶液(400 mL,200 mg ·L-1)反应不同时间段固相产物的TEM照片Fig. 4 TEM images of solid products from the oxidation of S2- (200 mg ·L-1,400 mL) by γ-MnOOH with initial pH 12.0 at 20 and 360 min in nitrogen atmosphere |
在上述反应过程中,200 mg ·L-1 Na2S溶液pH稳定在11.8左右,加入水锰矿后,pH变化不大,初始pH控制为12.0; 在氧化还原实验过程中,pH变化不太显著,经12 h反应后,pH稳定在11.3左右,可见,反应过程较为缓慢. 进一步调控反应体系初始pH,使模拟体系更接近自然环境,以期阐明水锰矿与水溶性硫化物的环境化学行为.
初始pH控制分别为10.0和8.0时,反应经过不足30 min,pH基本稳定在11.6左右,可见,水溶性硫化物初始氧化反应为释放OH-过程. 进一步对固相、 液相产物进行表征分析(图 5),对比初始pH为12.0的结果,明确初始pH对反应过程的影响.
 | 图 5 氮气环境下0.5 g水锰矿与S2-溶液(400 mL,200 mg ·L-1)不同初始pH反应过程S2-的氧化产物与转化率 Fig. 5 Solid/aqueous products and transformation rate of S2- from 200 mg ·L-1 S2- oxidized by γ-MnOOH in nitrogen atmosphere with initial pH 10 and pH 8 |
初始pH控制为10的体系中,反应进行10 min后有单质S生成,随着反应进行,γ-MnOOH衍射峰逐渐减弱,也即其含量降低,生成的Mn(OH)2在空气中也被氧化生成Mn3O4[图 5(a)]. 反应体系初始pH为8.0时,主要固相产物及变化趋势与pH为10.0的体系基本相同[图 5(c)],反应初期有单质S生成,且随反应进行单质S含量有上升的趋势. 对两反应体系液相产物及固相产物中单质S含量进行分析如图 5(b)、 5(d)所示. S32-氧化产物主要为S、 S2O2-、 SO32-和SO42-,且以单质S为主. 对比图 5和图 2可知,初始 pH控制为12.0、 10.0、 8.0,反应20 min后生成单质S的转化率分别为15.8%、 16.5%、 23.9%; 反应480 min后,初始 pH为12.0、 10.0、 8.0的体系中生成单质S的百分比均约82%,可知初始pH对水锰矿氧化硫化物的影响主要为S2-初始阶段的氧化速率. 在pH为12.0、 10.0和8.0的体系中,80%的S2-被氧化所需要的时间分别为45、 20和10 min,S2-的转化率变化趋势进一步说明随着pH升高,反应速率降低. 研究表明pH影响溶解硫的化学组分及锰氧化物表面位点进而影响水溶性硫化物的氧化速率[25].
调控体系初始pH为8-12,而在反应过程中,释放OH-使得体系酸碱度稳定范围为pH 11.0-12.0,进而对产物组分影响不大. 为了明确pH对反应动力学的影响,可恒定体系pH进行比较[9, 10]. 在缺氧条件下,反应体系初始pH分别控制为12.0、 10.0、 8.0,反应过程中pH均会快速上升至约为12.0且维持稳定,而初始pH越小,S2-初始氧化速率越快. 不同条件下反应产物及变化趋势基本一致,单质硫均为主要产物,固相产物主要为Mn(OH)2沉淀,反应过程可表述为:
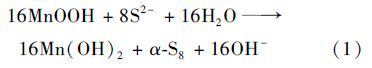
在表生环境中,氧气参与硫化矿物与Mn(Ⅱ)的氧化反应,为此,在上述体系中通入氧气以考察有氧环境中水锰矿与水溶性硫化物的反应过程,并对O2氧化硫化矿物的反应过程进行了研究. S2-浓度为200 mg ·L-1的溶液体系中,添加水锰矿后pH约为12.0,随着反应进行,pH基本稳定在11.8. 反应过程液相产物及浓度变化趋势如图 6所示.
 | 图 6 有氧氛围下0 g、0.5 g水锰矿与S2-溶液(400 mL,200 mg ·L-1)反应产物及S2-的转化率 Fig. 6 Transformation rate from S32- to S2O2-,SO32-,SO42- and the oxidation rate of S2- (200 mg ·L-1,400 mL) by single oxygen and γ-MnOOH in oxygen atmosphere |
O2与水溶性S32-反应10 h后,S2-转化率约为80.0%且仍有上升趋势[图 6(a)],且水溶性硫化物转化中间产物主要为 S2O2-和SO432-. 开放体系中,水锰矿与水溶性S2-反应90 min后,S2-转化率约为93.0%[图 6(b)],而后基本趋于稳定; 在封闭体系中通氮气条件下反应1.5 h,S2-转化率达99.7%(图 2). 有氧环境略降低了水锰矿氧化硫化物的反应速率. 水溶性硫化物转化中间产物主要为单质S和S2O2-,且单质S含量先上升到约38%后下降. S2O32-浓度呈上升趋势,10 h内其含量上升至60%左右. SO432-在反应初期含量上升,后基本维持在10%左右,可见氧气存在促进了高价态硫氧化物的生成,O2能进一步氧化S生成S2O2-和SO432-. 氧气较水锰矿具有更高的氧化活性,可溶性硫化物(S2-)氧化产物后期主要为高价态硫氧酸根离子. 氧气在水中溶解度有限,进而影响其扩散速率,因而较水锰矿粉末与水溶性硫化物反应速率慢,且氧气氧化水溶性硫化物主要为S2O2-和SO42-[9],使得与水锰矿反应的S2-浓度降低,因此,在有氧环境中S2-的总体转化率偏低.
在有氧环境中,水锰矿还原生成的Mn(OH)2沉淀会进一步氧化生成Mn3O4和β-MnOOH等. 在本实验中,固相产物X-射线衍射谱如图 7所示,可见,γ-MnOOH能保持较好的化学稳定性,经过约10 h反应,其晶体结构保持稳定; 反应40 min后出现了单质S的衍射峰,且初期特征衍射峰强度有增加的趋势,表明其含量增加,而后衍射峰逐渐消失,也即单质S的进一步氧化生成高价态的硫氧酸根离子. 这些与图 6所示结果较为一致.
 | 图 7 有氧环境下初始pH 12,水锰矿(0.5 g)与S2-溶液(400 mL,200 mg ·L-1)反应产物XRD图谱 Fig. 7 XRD patterns of solid products from the reaction of S2-(400 mL,200 mg ·L-1) and 0.5 g γ-MnOOH in the presence of oxygen with initial pH 12 |
在此过程发现有氧环境中,γ-MnOOH参与水溶性硫化物的氧化,利于S2O32-的生成,且维持很好的化学稳定性,即γ-MnOOH表现出良好的催化活性与稳定性. 为进一步明确水锰矿催化稳定性,TEM表征反应过程中固相产物微观形貌. 如图 8所示,有氧环境中,经过30 min和4 h反应,晶体微观形貌无显著变化,这与XRD分析结果一致,也即在有氧环境中水溶性硫化物的氧化,水锰矿表现出良好的催化稳定性. 而在相同条件下,钙锰矿出现明显的粉化现象[9],进一步说明氧化活性弱的水锰矿才能表现出催化稳定性.
上述开放体系中,O2加速了单质S的进一步氧化,维持了水锰矿的化学稳定性,水锰矿可催化氧气氧化水溶性硫化物生成S2O32-,具体反应如下:
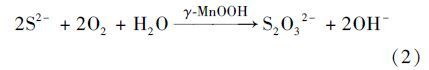
 | (a) 30 min; (b) 4 h图 8 有氧环境中水锰矿(0.5 g)与S2-溶液(200 mg ·L-1,400 mL)反应30 min和4 h产物的TEM照片 Fig. 8 TEM images of solid products from the oxidation of S2- (200 mg ·L-1,400 mL) by 0.5 g γ-MnOOH at 30 min and 4 h in the presence of oxygen with initial pH 12 |
(1)在缺氧的环境中,水锰矿氧化S32-可生成单质S、 S2O2-、 SO32-和SO42-,且以单质S为主要产物. 初始pH对反应过程影响较小,主要对S2-的转化率和单质S的生成速率有一定影响. 随着pH降低,S2-转化率和生成单质S的速率加快,而对长时间氧化产物组分影响较小.
(2)有氧环境体系中,氧气能将单质S进一步氧化为高价硫氧酸根离子,且水锰矿能保持较好的晶体结构和微观形貌,也即在表生有氧环境中,水锰矿表现出良好的催化特性.
| [1] | Hayes S M, Root R A, Perdrial N, et al. Surficial weathering of iron sulfide mine tailings under semi-arid climate[J]. Geochimica et Cosmochimica Acta, 2014, 141 : 240-257. |
| [2] | 卢信, 冯紫艳, 商景阁, 等. 不同有机基质诱发的水体黑臭及主要致臭物(VOSCs)产生机制研究[J]. 环境科学, 2012, 33 (9): 3152-3159. |
| [3] | 孙丽娟, 段德超, 彭程, 等. 硫对土壤重金属形态转化及植物有效性的影响研究进展[J]. 应用生态学报, 2014, 25 (7): 2141-2148. |
| [4] | Mongelli G, Sinisi R, Mameli P, et al. Ce anomalies and trace element distribution in Sardinian lithiophorite-rich Mn concretions[J]. Journal of Geochemical Exploration, 2015, 153 : 88-96. |
| [5] | Zhu M Q, Paul K W, Kubicki J D, et al. Quantum chemical study of arsenic (Ⅲ, Ⅴ) adsorption on Mn-oxides: Implications for arsenic (Ⅲ) oxidation[J]. Environmental Science & Technology, 2009, 43 (17): 6655-6661. |
| [6] | 徐建, 张莹, 李蕾, 等. δ-MnO2氧化降解苯酚的机理研究[J]. 环境科学学报, 2013, 33 (4): 1010-1016. |
| [7] | Li Y, Wei D B, Du Y G. Oxidative transformation of levofloxacin by δ-MnO2: Products, pathways and toxicity assessment[J]. Chemosphere, 2015, 119 : 282-288. |
| [8] | 李媛, 魏东斌, 杜宇国. 锰氧化物对有机污染物的转化机制研究进展[J]. 环境化学, 2013, 32 (7): 1288-1299. |
| [9] | Gao T Y, Shi Y, Liu F, et al. Oxidation process of dissolvable sulfide by synthesized todorokite in aqueous systems[J]. Journal of Hazardous Materials, 2015, 290 : 106-116. |
| [10] | Qiu G H, Li Q, Yu Y, et al. Oxidation behavior and kinetics of sulfide by synthesized manganese oxide minerals[J]. Journal of Soils and Sediments, 2011, 11 (8): 1323-1333. |
| [11] | 陈天虎, 汪家权. 天然锰矿物催化氧化法处理含硫废水研究[J]. 环境科学, 1993, 14 (4): 58-61. |
| [12] | Herszage J, dos Santos Afonso M. Mechanism of hydrogen sulfide oxidation by manganese (IV) oxide in aqueous solutions[J]. Langmuir, 2003, 19 (23): 9684-9692. |
| [13] | 李倩, 俞颖, 赵雅兰, 等. 锰钾矿氧化硫化物特性与动力学研究[J]. 环境科学, 2011, 32 (7): 2102-2108. |
| [14] | 李倩, 俞颖, 刘凡, 等. 水钠锰矿氧化硫化物的过程与动力学研究[J]. 岩石矿物学杂志, 2011, 30 (6): 1007-1013. |
| [15] | Nico P S, Zasoski R J. Mn(Ⅲ) center availability as a rate controlling factor in the oxidation of phenol and sulfide on δ-MnO2[J]. Environmental Science & Technology, 2001, 35 (16): 3338-3343. |
| [16] | Post J E. Manganese oxide minerals: Crystal structures and economic and environmental significance[J]. Proceedings of the National Academy of Sciences of the United States of America, 1999, 96 (7): 3447-3454. |
| [17] | Lefkowitz J P, Rouff A A, Elzinga E J. Influence of pH on the reductive transformation of birnessite by aqueous Mn(Ⅱ)[J]. Environmental Science & Technology, 2013, 47 (18): 10364-10371. |
| [18] | Chiu V Q, Hering J G. Arsenic adsorption and oxidation at manganite surfaces. 1. Method for simultaneous determination of adsorbed and dissolved arsenic species[J]. Environmental Science & Technology, 2000, 34 (10): 2029-2034. |
| [19] | Qin Y W, Pan G, Zhang M M, et al. Adsorption of zinc on manganite (gamma-MnOOH): particle concentration effect and adsorption reversibility[J]. Journal of Environmental Sciences (China), 2003, 16 (4): 627-630. |
| [20] | Weaver R M, Hochella Jr M F, Ilton E S. Dynamic processes occurring at the CrⅢaq-manganite (γ-MnOOH) interface: simultaneous adsorption, microprecipitation, oxidation/reduction, and dissolution[J]. Geochimica et Cosmochimica Acta, 2002, 66 (23): 4119-4132. |
| [21] | Qiu G H, Huang H, Genuino H, et al. Microwave-assisted hydrothermal synthesis of nanosized α-Fe2O3 for catalysts and adsorbents[J]. The Journal of Physical Chemistry C, 2011, 115 (40): 19626-19631. |
| [22] | Zhang W X, Yang Z H, Liu Y, et al. Controlled synthesis of Mn3O4 nanocrystallites and MnOOH nanorods by a solvothermal method[J]. Journal of Crystal Growth, 2004, 263 (1-4): 394-399. |
| [23] | Zhang Y C, Qiao T, Hu X Y. Preparation of Mn3O4 nanocrystallites by low-temperature solvothermal treatment of γ-MnOOH nanowires[J]. Journal of Solid State Chemistry, 2004, 177 (11): 4093-4097. |
| [24] | Wang W J, Yu J C, Xia D H, et al. Graphene and g-C3N4 nanosheets cowrapped elemental α-sulfur as a novel metal-free heterojunction photocatalyst for bacterial inactivation under visible-light[J]. Environmental Science & Technology, 2013, 47 (15): 8724-8732. |
| [25] | Yao W S, Millero F J. The rate of sulfide oxidation by δMnO2 in seawater[J]. Geochimica et Cosmochimica Acta, 1993, 57 (14): 3359-3365. |