 2016, Vol. 37
2016, Vol. 37



2. 江苏省大气环境监测与污染控制高技术研究重点实验室, 南京 210044;
3. 南京大学大气科学学院, 南京 210093
2. Jiangsu Key Laboratory of Atmospheric Environment Monitoring and Pollution Control, Nanjing 210044, China;
3. School of Atmospheric Science, Nanjing University, Nanjing 210093, China
随着我国工业化程度以及城市化进度加快,大气污染在诸多环境问题中尤为突出. 大气气溶胶能直接吸收和散射太阳辐射,影响能见度,进而造成灰霾等环境问题[1],同时影响整个地球系统太阳辐射的收支平衡,引起全球气候变化. 而且大气细颗粒物(PM2.5)可以随着呼吸进入人的肺泡,粒子所含有机物、 重金属等污染物会引起一系列健康问题. 大气气溶胶作为成云重要的云凝结核(cloud condensation nuclei,CCN)影响云滴浓度及云的寿命,直接或间接影响地球大气系统的辐射收支平衡,从而间接影响整个气候系统的变化. 由化石燃料和生物质的燃烧不完全产生的黑碳[2, 3](black carbon,BC),因其吸收阳光的能力较强[4],是全球变暖的重要角色[5]. BC从新鲜排放到老化的过程中,其外层通常会被水溶性无机盐[如(NH4)2SO4、 NH4NO3等]和水溶性有机物所包裹,因此老化的BC具有较好的亲水性,能够活化成长为CCN[6, 7, 8]. 气溶胶活化为云凝结核的能力(CCN活性)与其粒径及化学组分有密切的联系[9],有研究指出,粒径大小是影响气溶胶CCN活性的主要因素; 而其他一些研究表明气溶胶的化学组成也至关重要[10, 11, 12]. 实验表明,颗粒物表面覆盖有亲水性物质时,即使颗粒物核心为疏水性或者难溶性物质,其CCN活化能力将大大提高[13, 14, 15, 16, 17]. 而气溶胶粒子外表如果含有降低表面张力的活性剂,也可导致气溶胶CCN活性的增加[18, 19, 20]. 近年来,随着气溶胶对环境的影响日益加深,我国对气溶胶吸湿性、 混合状态及云凝结核的观测研究逐渐增多,在石家庄、 武清、 黄山、 上海、 南京,广州以及银川和四川资阳等地均进行了相关观测[21, 22, 23, 24, 25, 26, 27, 28, 29, 30]. 一般认为,气溶胶按吸湿性分为不吸湿、 弱吸湿和强吸湿三部分,不吸湿组分一般为新鲜排放的黑炭和不溶性有机物,弱吸湿部分为老化的黑炭,而强吸湿部分主要为水溶性无机物和小分子的水溶性有机物. 徐彬等[21]在南京北郊进行的气溶胶吸湿性观测表明,30-230 nm的气溶胶粒子吸湿性并不随粒径而变化. 而王轩[22]在北京进行的观测结果表明100、 150和200 nm颗粒物的吸湿生长因子高于25 nm和50 nm颗粒物,Liu等[23]在武清的观测也表明较大的颗粒物具有较高的吸湿性,这可能是由于南北区域大气颗粒物化学组分不同所引起. 颗粒物在大气中主要以两种混合态存在:内部混合和外部混合. 内混状态即BC由无机物与有机物均匀混合,颗粒物具有较为均一的化学组成和密度; 而外混状态指的是BC和其他无机物与有机物处于外接或者分离的状态,BC与其他颗粒物具有不同密度. Yin等[24]在上海对气溶胶混合态进行了观测,发现城市气溶胶基本都以内混状态存在. 目前国内对于云凝结核进行了较多观测研究,结果表明污染地区的云凝结核浓度一般较高,如京津冀[23]、 珠三角[25, 26]、 长三角[21]等区域云凝结核的平均浓度均在12 000个 ·cm-3以上,而西北西南区域[27, 28]的云凝结核浓度则相对较低,在6 000个 ·cm-3左右.
降水过程和灰霾天气对于CCN有很大的影响,南京近年来灰霾天气概率增加,在霾事件中,气溶胶粒子会显著影响地球表面和大气中的辐射平衡,并且通过改变CCN浓度和组成来间接影响气候[30]. 本研究于2014年9月11-18日在南京江心洲进行外场观测,分析了气溶胶特性及其混合态、 CCN活性变化,重点讨论了观测期间的灰霾和降水过程对气溶胶CCN活性和混合态的影响.
1 材料与方法 1.1 观测地点观测时间为2014年9月11-18日,观测地点位于江心洲南京市气象局(32.06°N,118.70°E),如图 1所示. 观测点距离江心洲东岸大约100 m. 奥体中心位于观测点东南方向,距离大约3 km. 该观测点具备规范化的气象要素观测设施,基础设施完善,周围交通源排放较少. 由于江心洲东岸为南京市自来水采水点,禁止航运通行,因此船舶航运相关排放不会对该处造成影响. 周边没有明显的污染源,因此具有较好的代表性.
 | 图 1 观测地点的地理位置示意 Fig. 1 Location of the observation site |
观测期间仪器布局如图 2所示,整个观测系统承载在一个可移动观测方舱内进行,室内温度由空调保持相对恒温,约25℃,进样口PM2.5切割头离地面约5 m. 整个系统分为两大部分,分别是分粒径气溶胶CCN活性测量系统和气溶胶有效密度测量系统. 采用中流量采样器(HY100型,青岛恒远)采集PM2.5气溶胶样品,采样流量为100 L ·min-1. 实时气象数据来自南京市气象局的自动气象监测站.
 | 图 2 仪器布局示意 Fig. 2 Setup of the instruments |
采用一台扫描电迁移率粒径谱仪(Scanning mobility particle sizer,SMPS,Model 3936,美国TSI)测量气溶胶粒径谱分布,时间分辨率为3 min. SMPS主要包括两个部分:差分迁移分析仪Differential mobility analyzer(DMA,Modal 3080,美国TSI)和凝结核计数器Condensation particle counter(CPC,Modal 3776,美国TSI).
CCN活性通过Droplet Measurement Technology(DMT,USA)生产的云凝结核计数器(CCNC)测量. 总流量为0.5 L ·min-1,鞘流样流比为10 ∶1,分别为0.45 L ·min-1和0.05 L ·min-1. 4个过饱和度(0.1%、 0.2%、 0.4%和0.8%)组成一个循环,除了过饱和度0.1%的时间为15 min,其他过饱和度的时间均为6 min,一共是31 min一个循环.
样品气流经主进样口先进入一个Nafion干燥管,相对湿度控制在30%以下[31]. 然后样流经过气溶胶中和器,使颗粒物达到正态分布带电. 带电气溶胶经过DMA进行单粒径输出,经校正后,实际输出粒径分别为76、 111、 138和181nm,单粒径气溶胶分为两路,一路(0.5 L ·min-1)进入CCNC进行CCN活性测量,另一路(0.3 L ·min-1)则进入CPC(Modal 3776)进行总气溶胶浓度(CN)的测量得到气溶胶单粒径活化率谱图,SMPS的鞘流样流比为10 ∶1.
在观测前后用(NH4)2SO4对CCNC进行了校准[32]. 利用Khler 公式将从(NH4)2SO4活化曲线得到的临界干粒径转化为实际过饱和度(effective supersaturation,SSe)[33]. 对温度梯度(temperature gradients,TG)和SSe进行线性拟合,可得到观测时设置过饱和度所对应的实际过饱和度. 校准结果表明,观测时实际过饱和度分别为0.14%、 0.23%、 0.41%和0.78%.
1.2.2 气溶胶有效密度测量系统观测气溶胶有效密度的系统主要包括4个部分:差分迁移分析仪DMA(Mode 3080),加热器,气溶胶质量分析仪(APM-3600,日本Kanomax Inc.)和凝结核计数器CPC(Mode 3772). 经过中和器的带电的气溶胶首先经过一台DMA进行单粒径输出,从DMA输出的单粒径气溶胶经过一个自行设计搭建的加热器:室温和300℃. 在每一个单粒径条件下通过三通阀控制气溶胶通过室温或300℃的加热管,随后分为两个气路,一路分别进入APM和CPC(Mode 3772)进行颗粒物质量的测定,测量某一粒径条件下不同温度的质量分布. 另一路进入分粒径气溶胶云凝结核活性测量系统,分别测量室温和300℃下的气溶胶活性. 在300℃下气溶胶颗粒物表面无机物和有机物基本全部挥发,剩下的可以近似等于BC的质量[34]. 颗粒物密度分布可由以下公式计算:
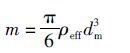
在观测实验开始之前,对DMA和APM用PSL(polystyrene latex)小球进行校准. 校准实验结果表明PSL小球粒径和密度的测量值与理论值吻合良好(误差<3%),重复校准实验随机误差小于1%,仪器状态良好.
1.2.3 加热器设计观测中,气溶胶颗粒物表面的无机物和有机物通过加热器(300℃)后去除. 加热器由2根1 m长的平行钢管组成,分别代表 2个温度环境:室温和300℃. 加热段的钢管由缠绕着保温带,由电热偶和一个温度控制器来加热和控制温度. 采样流量为1.1 L ·min-1,气溶胶在加热器中停留时间为1.7s. 样气经过加热管后,分别进入气溶胶云凝结核活性测量系统和有效密度测量系统. 以往研究表明,气溶胶经过加热器后,挥发蒸汽的重新冷凝附着可以忽略不计[35, 36, 37].
2 结果与讨论 2.1 气象条件南京位于长江下游,濒江临海,是我国重要的港口交通枢纽. 气象条件的变化可直接影响当地空气质量. 整个观测期间的气象数据(能见度、 总辐射强度、 温度、 相对湿度、 风向与风速)时间变化序列如图 3所示. 大部分气象参数呈现很明显的日循环变化趋势,观测期间平均温度为22.3℃,平均相对湿度为85%,平均风速为3.5m ·s-1,大部分情况下风速均小于5m ·s-1,最大风速为6m ·s-1,比较有利于污染物的集聚老化,不利于污染物的扩散[38]. 观测期间多为晴天,其中9月12-13日期间有一次降雨过程,9月16日为重度灰霾天气,能见度低.
 | 图 3 气象参数:温度、 相对湿度、 能见度、 辐射强度、 风向、 风速 Fig. 3 Meteorological parameters: temperature,relative humidity,visibility,radiation intensity,wind direction and wind speed |
整个观测期间盛行东风,只有9月13日降雨期间为北风,结合后向轨迹图(HYSPLIT NOAA模型)[39]发现,9月11-15日气团从朝鲜地区途经东部海上进入南京,主要为东北气团; 9月16-18日主要受北方气团的影响,主要源自辽宁渤海方向.
2.2 颗粒物有效密度及混合状态在整个观测期间,DMA单粒径输出的气溶胶粒径分别为76、 111、 138和181 nm,经加热器将单粒径气溶胶加热至300℃后,BC表面所吸附的挥发性有机物与无机物基本已挥发[31]. 观测期间各个粒径的气溶胶粒子所含BC质量比重如图 4所示,BC在76 nm粒子中所占质量百分比为5.4%,111 nm为10%,138 nm为10.7%,181 nm占6.7%,表明BC在颗粒物中主要分布在111-138 nm之间. BC质量比重整体比较低. Kuwata等[40]在日本东京观测气溶胶BC质量百分比可高达70%,主要原因可能是其在东京的观测地点接近交通要道,汽车排放尾气对BC贡献大,同时观测期间为冬季,有民用燃煤,而本研究的观测地点远离交通排放源,时处夏季,无民用燃煤现象. 同时,我国工业多用含硫煤炭,所排放BC附着有大量(NH4)2SO4、 NH4NO3、 NH4Cl等无机物,降低了BC在颗粒物中的质量分数[41]. 虽然颗粒物中所含黑碳质量比例较低,但是含黑碳的颗粒物数较多,76、 111、 138和181 nm颗粒物中,含黑碳颗粒物的质量分数分别为51%、 57%、 70%和59%,表明BC是大气中重要的凝结核,对大气颗粒物的数量有重要贡献.
 | 图 4 气溶胶颗粒物所含黑碳质量百分比 Fig. 4 Mass fraction of black carbon in aerosol particle |
颗粒物在大气中主要以两种形态存在:内部混合和外部混合. 含不同化学组分的颗粒物有不同的密度,可以根据以往的研究结果,以颗粒物密度来大致确定颗粒物的主要化学组成. 在BC的组成中,元素炭(element carbon,EC)为BC主要组成成分,EC的密度相对较高,范围在1.7-1.9g ·cm-3[42],但是由于BC的多孔和不规则松散结构使得新鲜排放的BC密度大约为0.1-0.6 g ·cm-3[41],同时也受颗粒物粒径大小、 硫化物等含量影响. 故在实际外场观测中,笔者认为密度小于1.0 g ·cm-3的质量峰为新鲜排放或轻微老化的BC[43].
图 5所示的是观测期间常温下76-181 nm的干粒径密度扫描. 利用Gaussian模型对测得的数据进行拟合,通过计算可得出各粒径颗粒物有效密度. 观测期间密度分布以单峰为主,76-181 nm粒径范围的城市气溶胶主要是由BC、 有机物、 吸湿性硫酸盐和硝酸盐混合而成[44],(NH4)2SO4和NH4NO3的密度为1.75 g ·cm-3左右. 图 5(a)是一个比较典型的单峰密度分布(9月15日14:00 76 nm),即颗粒物的内部混合形态,峰出现在密度为1.0-2.0 g ·cm-3之间,峰值出现在-1.45 g ·cm-3,表明颗粒物可能含有为硫酸盐(密度为1.75 g ·cm-3)和有机物(密度为1.2-1.5 g ·cm-3)[45]附着在黑碳核上的内混状态. 图 5(b)呈明显的双峰分布(2014年9月15日 09:00 120 nm),即外混状态. 主峰峰值为1.49 g ·cm-3,表明是大部分有机物和一部分硫酸盐; 同时主峰左边有一个比较小的峰,出峰位置为0.75 g ·cm-3,这与新鲜排放的黑炭或轻微部分老化的黑炭特征相吻合.
 | 图 5 颗粒物分粒径密度分布 Fig. 5 Typical density distributions for 76,111,138,and 181 nm particles |
观测期间气溶胶主要以内混状态为主,观测到的少数双峰外部混合状态出现在111 nm和138 nm,111 nm占40%,而138 nm占60%,BC的峰密度在0.5-1.0 g ·cm-3之间,这表明新鲜排放黑炭主要集中在138 nm左右. Levy等[43]在美国休斯敦观测到的新鲜排放的BC主要集中在150-240 nm,而本研究中150 nm以上粒径颗粒物并未观测到外混状态. 可能由于夏天光化学反应活跃,BC的老化速率较快,到达观测点的BC已经和硫酸盐、 硝酸盐、 有机物等充分混合,形成内混状态.
图 5中76 nm和181 nm颗粒物密度大约在2.3 g ·cm-3和3.3 g ·cm-3处有一个小峰,这可能是由于粒子带双电荷所引起的. 据报道,根据颗粒物形态,双电荷粒子经APM测量后,其密度大概为正常密度的1.3-1.7倍,主要原因为APM设计原理以粒子携带单电荷为前提,因而带多电荷粒子通过APM离心筛选后,测算出来的质量要比实际值偏大[46].
图 6表示的是观测期间不同粒径(76、 111、 138和181 nm)颗粒物密度随时间变化趋势图. 除了降雨期间密度呈明显下降趋势(蓝色阴影部分),其他时段颗粒物密度没有显著的波动. 76、 111、 138和181 nm平均密度分别为1.44、 1.46、 1.50和1.58 g ·cm-3,可以看出随着颗粒物粒径增大,密度也逐渐增大. 78、 111和138 nm粒径的颗粒物密度在1.21-1.64 g ·cm-3之间,表明颗粒物中可能含有(NH4)2SO4和NH4NO3等无机物,与有机物以不同比例内混. 而181 nm粒径颗粒物密度范围在1.53-1.63 g ·cm-3之间,说明颗粒物以(NH4)2SO4、 NH4NO3等无机物为主,并含有少量的有机物.
 | 图 6 气溶胶颗粒物(76、 111、 138和181 nm)有效密度时间序列 Fig. 6 Temporal variation of the peak aerosol effective densities for 76,111,138,and 181 nm particles |
由图 3可以看出,9月16日能见度低,为重度霾天. 结合后向轨迹图[图 7(a)],晴天时,气团来自日本海,绕过朝鲜半岛经东海进入南京,空气相对洁净; 而控制霾天的气团来自于韩国忠清南道工业区,此工业区以汽车、 石油产品、塑料、 纸产品、 钢铁板材、 石油化学合成原料等工业为主. 大气能见度从9月16日00:00开始下降,09:00达到最低值,在此期间,颗粒物密度有所上升. 将中流量采样器(HY100型,青岛恒远)采得的PM2.5样品经离子色谱进行分析,颗粒物的化学组分如图 7(b)所示,图中分别展示了晴天与霾天颗粒物的化学组成,与晴天相比,霾天有机物含量由48%降低至33%,而SO42-和NH4+却有明显的增长,因此霾天颗粒物密度明显大于晴天颗粒物密度.
 | 图 7 9月16日的大气气团后向轨迹分析及晴天和霾天化学组分质量百分比 Fig. 7 Back-trajectories analysis on September 16 and chemical composition on clean day and hazy day |
图 8表示了气溶胶颗粒物在经过300℃高温加热过后,表面有机物与(NH4)2SO4、 NH4NO3等无机物挥发后,剩下的颗粒物质量主要为BC. 图 8(a)展示的是9月11日观测到的一组数据,在天气晴朗的情况下,在300℃条件下挥发的有机物与无机物约占92%,BC所占比例为8%. 图 8(b)是9月12日的一组数据,经过9月12日的降雨过程,BC在颗粒物中所占比例增加至13%,可能是由于在降雨过程中,雨水的冲刷会使颗粒物浓度下降,导致降雨时(后)新鲜排放的BC占空气中悬浮颗粒物的相对比例增高,颗粒物密度下降.
 | 图 8 气溶胶颗粒物加热前后质量分布 Fig. 8 Aerosol particle mass distribution before and after heating |
根据CCNC和CPC所得数据分别对几个特定粒径(76、 111、 138和181 nm)在给定某一过饱和度下的活化率进行计算,再对同一粒径下的活化率以sigmoidal函数进行拟合,拟合结果如图 9所示. 在某一粒径下活化率为50%时的过饱和度为临界过饱和度(SSc),可以认为某一个粒径下的颗粒物在过饱和度大于SSc时完全活化. 由图 9可知,不同粒径的拟合曲线差别明显. 在观测期间76、 111、 138 和181 nm颗粒物的SSc分别为0.25、 0.13、 0.06和0.015. 对于小粒径颗粒物(76 nm)在过饱和度小于0.1%时几乎不活化,而对于大颗粒物(138和181 nm)在SSc低于0.1时就趋近于全部活化. 在图 9中除了76 nm最终活化率没有达到1,大颗粒物活化率均接近全部活化,说明颗粒物内混了一些高吸湿性的物质.
 | 图 9 观测期间颗粒物(76、 111、 138和181 nm)云凝结核活性曲线 Fig. 9 CCN activation curves of particles (76,111,138,and 181 nm) during the observation period |
图 10分别展示了颗粒物在晴天、 霾天和雨天的CCN活化谱图和颗粒物分粒径临界过饱和度. 从图 10(d)可以看出整体趋势是颗粒物临界过饱和度随着粒径的增大而逐渐减小. 晴天时,76、 111、 138和181 nm临界过饱和度分别为0.25%、 0.12%、 0.08%和0.02%; 而在雨天,颗粒物的临界过饱和度最高,分别为0.26%、 0.14%、 0.07%和0.03%; 霾天最低,临界过饱和度降低为0.22%、 0.12%、 0.05%和0.02%,说明霾天颗粒物最易活化为CCN,雨天颗粒物较难活化. 这与上文中气溶胶密度与化学成分的观测结果一致,霾天颗粒物中无机盐含量相对较高,而雨天BC含量相对增加. 同时,由图 10(d)可以看出,在气溶胶颗粒物粒径较小时,雨天、 晴天和霾天的临界过饱和度依次降低,较为明显; 而在颗粒物粒径较大时,其临界过饱和度差距不大. 这是由于颗粒物在粒径较小时,其化学组分对颗粒物的云凝结核活性影响较大,而在粒径较大的时候,颗粒物已经充分老化,化学成分接近,因此此时影响颗粒物云凝结核活性的主要因素为颗粒物粒径. 晴天、 雨天和霾天的κ平均值分别为0.37、 0.29、 0.39,κ值越大,吸湿性越强. 整个观测期间κ平均值为0.35,这与珠三角的观测值(0.34±0.11)[47]基本一致,但比北京的测量值(0.31±0.08)[17]及文献[48, 49]中建议的内陆地区的κ值(0.3)略高,表明本研究观测点气溶胶的吸湿性较强.
 | 图 10 晴天、 霾天和雨天云凝结核活性曲线和临界过饱和度对干粒径曲线 Fig. 10 CCN activation curves and curves of critical supersaturation vs. particle size on clear,hazy and rainy days |
(1)观测期间大气颗粒物主要以内混状态存在,76、 111、 138和181 nm颗粒物的平均密度分别为1.44、 1.46、 1.50和1.58 g ·cm-3. 在少数情况下观测到颗粒物的外混状态,主要集中在111 nm和138 nm.
(2)BC在76、 111、 138和181 nm颗粒物中所占的质量分数分别为5.4%、 10.0%、 10.7%和6.6%,BC主要集中在111 nm和138 nm之间,整体含量较低,但是含有黑碳核的气溶胶颗粒物分别占51%、 57%、 70%和59%,表明BC是大气中重要的凝结核,对大气颗粒物的数量有重要贡献.
(3)76、 111、 138和181 nm颗粒物的临界过饱和度分别为0.25%、 0.13%、 0.06%和0.015%. 气溶胶颗粒物在晴天、 雨天、 霾天的吸湿性系数κ分别为0.37、 0.29、 0.39,化学组成对气溶胶颗粒物的云凝结核活性有较大影响.
| [1] | US Environmental Protection Agency (US EPA). How air pollution affects the view[R]. EPA-456/F-06e001, 2006. |
| [2] | Novakov T, Ramanathan V, Hansen J E, et al. Large historical changes of fossil-fuel black carbon aerosols[J]. Geophysical Research Letters, 2003, 30 (6): 1324, doi: 10.1029/2002GL016345. |
| [3] | Ito A, Penner J E. Historical emissions of carbonaceous aerosols from biomass and fossil fuel burning for the period 1870-2000[J]. Global Biogeochemical Cycles, 2005, 19 (2): GB2028, doi: 10.1029/2004GB002374. |
| [4] | Ramanathan V, Carmichael G. Global and regional climate changes due to black carbon[J]. Nature Geoscience, 2008, 36 (1): 335-358. |
| [5] | Jacobson M Z. Strong radiative heating due to the mixing state of black carbon in atmospheric aerosols[J]. Nature, 2001, 409 (6821): 695-697. |
| [6] | Lammel G, Novakov T. Water nucleation properties of carbon black and diesel soot particles[J]. Atmospheric Environment, 1995, 29 (7): 813-823. |
| [7] | Petzold A, Gysel M, Vancassel X, et al. On the effects of organic matter and sulphur-containing compounds on the CCN activation of combustion particles[J]. Atmospheric Chemistry and Physics, 2005, 5 (12): 3187-3203. |
| [8] | Dusek U, Reischl G P, Hitzenberger R. CCN activation of pure and coated carbon black particles[J]. Environmental Science & Technology, 2006, 40 (4): 1223-1230. |
| [9] | Zhang Q, Jimenez J L, Canagaratna M R, et al. Ubiquity and dominance of oxygenated species in organic aerosols in anthropogenically-influenced Northern Hemisphere midlatitudes[J]. Geophysical Research Letters, 2007, 34 (13): L13801, doi: 10.1029/2007gl029979. |
| [10] | Petters M D, Kreidenweis S M. A single parameter representation of hygroscopic growth and cloud condensation nucleus activity[J]. Atmospheric Chemistry and Physics, 2007, 7 (8): 1961-1971. |
| [11] | Lambe A T, Onasch T B, Massoli P, et al. Laboratory studies of the chemical composition and cloud condensation nuclei (CCN) activity of secondary organic aerosol (SOA) and oxidized primary organic aerosol (OPOA)[J]. Atmospheric Chemistry and Physics, 2011, 11 (17): 8913-8928. |
| [12] | Wong J P S, Lee A K Y, Slowik J G, et al. Oxidation of ambient biogenic secondary organic aerosol by hydroxyl radicals: effects on cloud condensation nuclei activity[J]. Geophysical Research Letters, 2011, 38 (22): L22805. |
| [13] | Chang R Y W, Liu P S K, Leaitch W R, et al. Comparison between measured and predicted CCN concentrations at Egbert, Ontario: Focus on the organic aerosol fraction at a semi-rural site[J]. 2007, 41 (37): 8172-8182. |
| [14] | Raymond T M, Pandis S N. Cloud activation of single-component organic aerosol particles[J]. Journal of Geophysical Research, 2002, 107 (D24): AC16-1-AAC16-8, doi: 10.1029/2002JD002159. |
| [15] | Raymond T M, Pandis S N. Formation of cloud droplets by multicomponent organic particles[J]. Journal of Geophysical Research, 2003, 108 (D15): 4469, doi: 10.1029/2003JD003503. |
| [16] | Broekhuizen K, Kumar P P, Abbatt J P D. Partially soluble organics as cloud condensation nuclei: role of trace soluble and surface active species[J]. Geophysical Research Letters, 2004, 31 (1): L01107, doi: 10.1029/2003GL018203. |
| [17] | Bilde M, Svenningsson B. CCN activation of slightly soluble organics: the importance of small amounts of inorganic salt and particle phase[J]. Tellus B, 2004, 56 (2): 128-134. |
| [18] | Kuwata M, Kondo Y, Miyazaki Y, et al. Cloud condensation nuclei activity at Jeju Island, Korea in spring 2005[J]. Atmospheric Chemistry and Physics, 2008, 8 (11): 2933-2948, doi: 10.5194/acp-8-2933-2008. |
| [19] | Moore R H, Ingall E D, Sorooshian A, et al. Molar mass, surface tension, and droplet growth kinetics of marine organics from measurements of CCN activity[J]. Geophysical Research Letters, 2008, 35 (7): L07801, doi: 10.1029/2008GL033350. |
| [20] | Padró L T, Tkacik D, Lathem T, et al. Investigation of cloud condensation nuclei properties and droplet growth kinetics of the water-soluble aerosol fraction in Mexico City[J]. Journal of Geophysical Research, 2010, 115 (D9): D09204, doi: 10.1029/2009JD013195. |
| [21] | 徐彬, 张泽锋, 李艳伟, 等. 南京北郊春季气溶胶吸湿性分析[J]. 环境科学, 2015, 36 (6): 1911-1918. |
| [22] | 王轩. 气溶胶吸湿特性研究[D]. 北京: 中国环境科学研究院, 2010. |
| [23] | Liu P F, Zhao C S, Göbel T, et al. Hygroscopic properties of aerosol particles at high relative humidity and their diurnal variations in the North China Plain[J]. Atmospheric Chemistry and Physics, 2011, 11 (7): 3479-3494. |
| [24] | Yin Z, Ye X N, Jiang S Q, et al. Size-resolved effective density of urban aerosols in Shanghai[J]. Atmospheric Environment, 2015, 100 : 133-140. |
| [25] | Tan H B, Yin Y, Gu X F, et al. An observational study of the hygroscopic properties of aerosols over the Pearl River Delta region[J]. Atmospheric Environment, 2013, 77 : 817-826. |
| [26] | 韩冰雪, 张国华, 毕新慧, 等. 广州城区夏季大气颗粒物数浓度谱分布特征[J]. 环境科学研究, 2015, 28 (2): 198-204. |
| [27] | 赵永欣, 牛生杰, 吕晶晶, 等. 2007年夏季我国西北地区云凝结核的观测研究[J]. 高原气象, 2010, 29 (4): 1043-1049. |
| [28] | 陈晨, 胡敏, 吴志军, 等. 四川乡村点新粒子生成特征及其对云凝结核数浓度的贡献[J]. 中国环境科学, 2014, 34 (11): 2764-2772. |
| [29] | 李琦, 银燕, 顾雪松, 等. 南京夏季气溶胶吸湿增长因子和云凝结核的观测研究[J]. 中国环境科学, 2015, 35 (2): 337-346. |
| [30] | Chung C E, Ramanathan V, Kiehl J T. Effects of the South Asian absorbing haze on the Northeast Monsoon and surface-air heat exchange[J]. Journal of Climate, 2002, 15 (17): 2462-2476. |
| [31] | Leng C P, Cheng T T, Chen J M, et al. Measurements of surface cloud condensation nuclei and aerosol activity in downtown Shanghai[J]. Atmospheric Environment, 2013, 69 : 354-361. |
| [32] | Rose D, Gunthe S S, Mikhailov E, et al. Calibration and measurement uncertainties of a continuous-flow cloud condensation nuclei counter (DMT-CCNC): CCN activation of ammonium sulfate and sodium chloride aerosol particles in theory and experiment[J]. Atmospheric Chemistry and Physics, 2008, 8 (5): 1153-1179, doi: 10.5194/acp-8-1153-2008. |
| [33] | Pruppacher H R, Klett J D. Microphysics of clouds and precipitation[M]. Dordrecht Boston: Kluwer Academic Publishers, 1997. 954. |
| [34] | Kondo Y, Komazaki Y, Miyazaki Y, et al. Temporal variations of elemental carbon in Tokyo[J]. Journal of Geophysical Research, 2006, 111 (D12): D12205, doi: 10.1029/2005JD006257. |
| [35] | Kondo Y, Sahu L, Kuwata M, et al. Stabilization of the mass absorption cross section of black carbon for filter-based absorption photometry by the use of a heated inlet[J]. Aerosol Science and Technology, 2009, 43 (8): 741-756. |
| [36] | Kuwata M, Kondo Y, Mochida M, et al. Dependence of CCN activity of less volatile particles on the amount of coating observed in Tokyo[J]. Journal of Geophysical Research, 2007, 112 (D11): D11207, doi: 10.1029/2006JD007758. |
| [37] | Wehner B, Philippin S, Wiedensohler A. Design and calibration of a thermodenuder with an improved heating unit to measure the size-dependent volatile fraction of aerosol particles[J]. Journal of Aerosol Science, 2002, 33 (7): 1087-1093. |
| [38] | Lei W F, Zhang R Y, Tie X X, et al. Chemical characterization of ozone formation in the Houston-Galveston area: A chemical transport model study[J]. Journal of Geophysical Research, 2004, 109 (D12): D12301, doi: 10.1029/2003JD004219. |
| [39] | Draxler R R, Hess C D. Description of the HYSPLIT_ 4 modeling system[R]. NOAA Technical Memorandum ERL ARL-224, Maryland: Silver Spring, 1997. |
| [40] | Kuwata M, Kondo Y, Takegawa N. Critical condensed mass for activation of black carbon as cloud condensation nuclei in Tokyo[J]. Journal of Geophysical Research, 2009, 114 (D20): D20202, doi: 10.1029/2009JD012086. |
| [41] | Zhang R Y, Khalizov A F, Pagels J, et al. Variability in morphology, hygroscopicity, and optical properties of soot aerosols during atmospheric processing[J]. Proceedings of the National Academy of Sciences of the United States of America, 2008, 105 (30): 10291-10296. |
| [42] | Bond T C, Bergstrom R W. Light absorption by carbonaceous particles: an investigative review[J]. Aerosol Science and Technology, 2006, 40 (1): 27-67, doi: 10.1080/02786820500421521. |
| [43] | Levy M E, Zhang R Y, Khalizov A F, et al. Measurements of submicron aerosols in Houston, Texas during the 2009 SHARP field campaign[J]. Journal of Geophysical Research, 2013, 118 (18): 10518-10534. doi: 10.1002/jgrd.50785. |
| [44] | Hu M, Peng J F, Sun K, et al. Estimation of size-resolved ambient particle density based on the measurement of aerosol number, mass, and chemical size distributions in the winter in Beijing[J]. Environmental Science & Technology, 2012, 46 (18): 9941-9947. |
| [45] | Kostenidou E, Pathak R K, Pandis S N. An algorithm for the calculation of secondary organic aerosol density combining AMS and SMPS data[J]. Aerosol Science and Technology, 2007, 41 (11): 1002-1010, doi: 10.1080/02786820701666270. |
| [46] | Tajima N, Sakurai H, Fukushima N, et al. Design considerations and performance evaluation of a compact aerosol particle mass analyzer[J]. Aerosol Science and Technology, 2013, 47 (10): 1152-1162. |
| [47] | Rose D, Nowak A, Achtert P, et al. Cloud condensation nuclei in polluted air and biomass burning smoke near the mega-city Guangzhou, China-Part 1: size-resolved measurements and implications for the modeling of aerosol particle hygroscopicity and CCN activity[J]. Atmospheric Chemistry and Physics, 2010, 10 (7): 3365-3383. |
| [48] | Gunthe S S, Rose D, Su H, et al. Cloud condensation nuclei (CCN) from fresh and aged air pollution in the megacity region of Beijing[J]. Atmospheric Chemistry and Physics, 2011, 11 (21): 11023-11039. |
| [49] | Jurányi Z, Gysel M, Weingartner E, et al. A 17 month climatology of the cloud condensation nuclei number concentration at the high alpine site Jungfraujoch[J]. Journal of Geophysical Research, 2011, 116 (D10): D10204, doi: 10.1029/2010JD015199. |