 2016, Vol. 37
2016, Vol. 37

2. 苏州科技学院环境生物技术研究所, 苏州 215009
 , HUANG Yong1,2
, HUANG Yong1,2 , LIU Xin1,2, YUAN Yi1,2, LI Xiang1,2, WANGYAN De-qing1,2, DING Liang1,2, SHAO Jing-wei1,2, ZHAO Rong1,2
, LIU Xin1,2, YUAN Yi1,2, LI Xiang1,2, WANGYAN De-qing1,2, DING Liang1,2, SHAO Jing-wei1,2, ZHAO Rong1,2
2. Institute of Environmental Biotechnology, Suzhou University of Science and Technology, Suzhou 215009, China
氮素排入水体会引起富营养化. 其中,硝酸盐在缺氧条件下会被转化为具有“三致”作用的亚硝盐,威胁人类健康. 氮素污染已成为公众关注的焦点.
众多脱氮技术中,厌氧氨氧化(anaerobic ammonium oxidation,ANAMMOX)是目前公认的最具前景的新型脱氮技术. 其以氨氮为电子供体,以亚硝酸盐为电子受体,在厌氧氨氧化微生物的作用下,两者反应生成氮气[1]. 该过程产泥率低,相比传统硝化反硝化脱氮可节约62%的能耗[2],目前已逐渐流行于荷兰、 德国、 奥地利、 瑞士等地[3, 4].
生物反硝化工艺的研发应用也是目前脱氮工艺的研究热点. 传统反硝化效果较好,但需要有机物的参与,其占地、 曝气能耗、 药剂投加及产泥量均较大. 氢自养反硝化产物为水和氮气,但氢自养反硝化微生物驯化培养不易,脱氮效果受氢气传质速率影响较大,且氢气制备过程较为复杂,氢气存储还涉及防爆安全问题[5, 6],故该工艺目前仅停留在实验室阶段.
单质硫自养反硝化工艺的处理效率较高,运行费用及污泥产率均较低[7, 8]. 但该反应碱度消耗量大,反应最终会产生大量硫酸盐,这在增加水中盐度的同时,也会带来水体发黑变臭的风险[9]. 为控制硫自养反硝化过程中碱度过度消耗导致的pH下降问题,有学者引入了石灰石[10, 11, 12],将其与单质硫颗粒按一定比例混合制成固定床,利用石灰石的碱性来补充一定的碱度. 但该过程中石灰石释放缓慢,其碱度补充程度往往赶不上消耗的速率,宏观上水体pH仍会较快地下降. 同时,石灰石所释放出的Ca2+会提高水的硬度[13]. 也有学者采用异养和自养反硝化混合体系,通过异养反硝化产生的碱度来缓解硫自养反硝化的耗碱问题. 同时,该过程中存在竞争硝酸盐,可以抑制自养反硝化的硫酸盐产生量[14, 15, 16]. 但该过程中有机物的投加会增加运行费用,且系统中异养反硝化的引入带来了污泥增长加快、 出水携带有机物等问题[17].
单质硫自养反硝化过程中,亚硝酸盐是反应的中间产物,但是在特定条件下由于亚硝酸盐转化速率受到限制,亚硝酸盐容易成为终产物[18, 19]. 若单质硫反硝化体系内同时存在ANAMMOX微生物和氨时,可以控制内部自养反硝化反应至亚硝酸盐阶段,接着通过厌氧氨氧化反应去除亚硝酸盐. 这样做,一方面将自养反硝化过程限定于产生亚硝酸盐阶段,一方面可以减少单质硫的投加量、 硫酸盐的产生量和碱度的消耗量; 另一方面厌氧氨氧化反应本身会产生碱度,可以补充部分自养反硝化的耗碱量. 基于此原理可见,实际运行中可以采用连续流反应器或序批式进行耦合反应. 本研究拟将硫自养反硝化与ANAMMOX反应进行耦合,考察引入厌氧氨氧化过程后,其对硫自养反硝化过程中硫酸盐产生、 碱度消耗及pH值的影响,以明确两者之间协同脱氮的反应条件.
1 材料与方法 1.1 连续流实验装置采用全混式厌氧搅拌罐,其有效体积2.0 L,直径为φ100 mm,有效高度为250 mm,设置固液分离器. 由蠕动泵从底部进水,出水经固液分离器进行水、 固分离,最后经出水堰排水. 反应器出水处、 搅拌器接口处均设置水封装置,并且进水采用氮气吹脱除氧,以确保反应器处于厌氧状态. 产生氮气通过固液分离装置由上部水封装置自然溢出. 反应器温度控制为35℃±0.5℃,搅拌强度120 r·min-1. 为避免光照对厌氧氨氧化菌的伤害,反应器外部用黑色遮光布覆盖避光. 反应接种200 g(湿重)厌氧氨氧化颗粒污泥,并添加100 g粒径为8~12目的单质硫颗粒. 实验装置如图 1 所示.
 | 图 1 连续流装置示意 Fig. 1 Scheme of experimental reactor for continuous flow |
反应启动阶段通过减少水力停留时间(HRT为16、 8、 4 h)和适当提高进水硝态氮浓度(100 mg·L-1,200mg·L-1)来培养硫自养反硝化微生物. 反应器稳定后,进水加入一定量的氨氮(60mg·L-1),实现同步硫自养反硝化和厌氧氨氧化,考察添加氨后实现部分厌氧氨氧化对硫自养反硝化处理硝酸盐废水过程中产生硫酸盐的变化,消耗碱度的变化,及pH值的变化.
1.2 批试实验取1.0 g培养完毕的单质硫反硝化污泥于50 mL血清瓶内,加入0.5 g 12~18目硫颗粒和50 mL含100 mg·L-1 NH4+-N和200 mg·L-1 NO3--N的营养液,高纯氮吹脱20 min,并迅速密封反应10 h. 采用磁力搅拌器控制搅拌转速为160、 290、 320、 400、 500、 600和700 r·min-1,通过HCl和NaOH调节pH值为5.08、 6.19、 6.90、 8.08、 8.91,分别考察搅拌强度和pH值对耦合脱除氨和硝酸盐反应的影响.
1.3 接种污泥及配水实验用单质硫为8~12目,投放反应器前首先将单质硫颗粒放入筛网中来回振荡5 min以去除细小单质硫颗粒. 反应器接种厌氧氨氧化污泥取自本课题组培养亚硝酸盐型厌氧氨氧化反应器,其颗粒直径1~2 mm,原反应器氮负荷达到2.5 kg·(m3·d)-1. 基本培养液成分为(mg·L-1): KH2PO4 5、 CaCl2·2H2O 136、 MgCl2·7H2O 200、 NaHCO3=KHCO3=500,微量元素Ⅰ 1 mL·L-1,微量元素Ⅱ 1.25 mL·L-1. 微量元素Ⅰ组成:EDTA 5 000 mg·L-1,硫酸亚铁5 000 mg·L-1. 微量元素Ⅱ的组成如表 1所示.
| 表 1 微量元素Ⅱ 的组成 Table 1 Composition of trace element Ⅱ |
实验中各污染物指标的监测方法均参照文献[20]进行:NH4+-N采用纳氏试剂分光光度法,NO2--N采用N-(1-萘基)-乙二胺分光光度法、 NO3--N采用紫外分光光度法、 总铁采用邻菲啰啉分光光度法、 SO42--S采用铬酸钡分光光度法、 pH值采用pHS-3TC型酸度计测定.
2 结果与分析全混式厌氧搅拌罐内首先接种课题组已前期培养出的厌氧氨氧化污泥,并加入单质硫颗粒. 启动初期采用只含有NO3-的无机营养液富集硫自养反硝化微生物,并通过缩短停留时间和提高进水硝酸盐浓度逐步提升氮负荷. 当反应器获得稳定的硫自养反硝化能力后,反应器内加了适量的氨,使得反应器内发生厌氧氨氧化反应,考察硫自养反硝化和厌氧氨氧化联合的优势. 其中0~3 d,停留时间为16 h,进水硝态氮浓度为100mg·L-1; 4~10 d停留时间为8 h,进水硝态氮浓度为100mg·L-1; 11~15 d,水力停留时间为4 h,进水硝态氮浓度为100mg·L-1; 16~30 d,水力停留时间为5.3 h,进水硝态氮浓度为200mg·L-1; 31~59 d,水力停留时间为5.3 h,进水硝态氮为200mg·L-1,进水氨氮为60mg·L-1. 整个反应过程中反应器保持搅拌强度为120 r·min-1,温度控制为35℃±0.5℃,进水pH值8.0~8.5.
2.1 硫自养反硝化反应启动特性如图 2(a)和图 2(d)所示,开始前3 d,当停留时间为16 h,进水NO3--N为100mg·L-1左右,出水NO3--N浓度低于5mg·L-1,出水SO42--S为70~120mg·L-1. 生成硫酸盐与硝酸盐摩尔损失比为0.15~0.85,该实际值远小于硫自养反硝化反应理论硫氮比的1.1[18, 21, 22, 23],并且出水TOC高于进水TOC,出水IC高于进水IC(进水平均TOC 3.7 mg·L-1,IC 112.4 mg·L-1,出水平均TOC 25.3 mg·L-1,IC 124.5 mg·L-1),说明启动初期系统内由于原有微生物没有适应进水反应环境,大量微生物死亡释放了内碳源引起异养反硝化脱氮[24, 25]. 4~10 d,停留时间缩短至8 h,进水NO3--N为100mg·L-1左右,此阶段由于停留时间的缩短,出水硝态氮升高至50 mg·L-1,但随后又下降至20mg·L-1. 出水硫酸盐也呈先下降再上升的趋势,其浓度为183.4~228.5mg·L-1. 此阶段硫氮摩尔比为0.85~1.31,可知该阶段微生物逐渐适应了进水水质,硫自养反硝化反应速率进一步提高. 11~15 d,将水力停留时间减少至4 h,进水NO3--N为100mg·L-1,出水NO3--N逐渐降低至20mg·L-1,此时出水SO42--S升高至228.1~293.4 mg·L-1,反应的硫氮摩尔比为1.20~1.24,比例与理论值1.1较为接近,说明反应器内脱氮主要由硫自养反硝化反应完成,此时反应器容积负荷已达到0.16~0.49 kg·(m3·d)-1. 16~30 d,保持水力停留时间为5.3 h,并增加进水NO3--N至200mg·L-1,该阶段内,出水NO3--N由上升至72.3mg·L-1,后逐步降低至50mg·L-1,去除负荷稳定在0.56~0.71kg·(m3·d)-1. 说明接种ANAMMOX污泥可在30 d快速启动硫自养反硝化反应器.
 | 图 2 硫自养反硝化反应启动过程中各底物变化 Fig. 2 Variation of substrates during the starting up period of the sulfur-based autotrophic denitrification |
如图 2(b)和图 2(c)所示,整个启动过程中,由于反应器接种ANAMMOX污泥,出水氨氮和亚硝氮均小于2.0mg·L-1. 当前IC为100~130mg·L-1条件下,反应过程中pH值由8.0~8.4降低至6.5~7.5. 当进水NO3--N为100mg·L-1左右时,出水pH值大于7.10~7.45; 当进水NO3--N为200mg·L-1左右时,出水pH值为6.4~6.9. 该反应为消耗IC过程. 由于硫自养反硝化为消耗碱度过程,当硫自养反硝化微生物逐步富集后,体系内IC消耗量随硝酸盐消耗量的增加而增加. 进水NO3--N为200mg·L-1左右时,出水IC值已降低至30~50mg·L-1.
2.2 耦合硫自养反硝化和厌氧氨氧化反应特性当反应器运行至第31 d后开始,进水中均添加了60mg·L-1氨氮. 如图 3所示,在第31~37 d控制进水NO3--N为175mg·L-1. 结果出水SO42--S由432.3mg·L-1降低至292.8mg·L-1,出水NO3--N稳定45.3~48.2 mg·L-1,此时NH4+-N由60 mg·L-1降低至10.1~19.2 mg·L-1. 该阶段消耗单位硝酸盐产生的硫酸盐的量,即为Δn(SO42-)∶Δn(NO3-),由1.2降低至0.9,出水pH值由6.5上升至7.2,出水IC由39.8 mg·L-1上升至54.2 mg·L-1. 运行过程中出水亚硝酸盐始终低于3.4 mg·L-1,硝态氮去除负荷仍维持在0.57~0.71 kg·(m3·d)-1. 说明添加60mg·L-1氨氮后对硫自养反硝化反应无显著抑制作用,并且由于耦合了厌氧氨氧化反应,出水硫酸盐更低,消耗碱度更少.
第37~59 d,进水NO3--N为200 mg·L-1左右,出水NO3--N稳定在50.2~57.3 mg·L-1,出水SO42--S保持在344.2~414.8 mg·L-1,出水NH4+-N为10.6~21.2 mg·L-1. 该阶段Δn(SO42-)∶Δn(NO3-)为0.94~1.02,出水pH值为7.06~7.25,出水IC由46.2~54.4 mg·L-1,硝态氮去除负荷为0.66~0.88 kg·(m3·d)-1. 反应体系各指标保持相对恒定,说明该系统运行已稳定.
 | 图 3 添加氨氮后耦合硫自养反硝化和厌氧氨氧化反应各底物变化 Fig. 3 Variation of substrates during the period of simultaneous sulfur-based denitrification and anaerobic ammonia oxidation after addition of ammonia |
由于主体反应单质硫自养反硝化为固-液生物反应并且耦合反应涉及两大类菌群的联合,故一方面考察搅拌强度主导的固-液传质对耦合反应的影响,另一方面考察pH值调节两类菌群活性而导致的对耦合反应的影响. 搅拌强度以GT值表示,因转速与搅拌强度存在正相关关系,本实验采用序批试实验的方法,分别控制反应体系搅拌强度为160、 290、 320、 400、 500、 600、 700 r·min-1(采用GT值表示为22、 55、 64、 89、 125、 165 s-1)及pH值为5.08、 6.19、 6.90、 8.08、 8.91,并保持其他条件不变(温度35℃、 基本培养液不变、 初始NO3--N 200mg·L-1、 初始NH4+-N 100 mg·L-1)的情况下反应8 h,考察搅拌强度和pH值对耦合反应脱氮能效的影响.
控制体系初始pH值为7.8,搅拌强度对耦合反应的影响如图 4(a)所示. 当搅拌强度为160~700 r·min-1(GT值为22~165 s-1)时,NO3--N的转化速率由8.75mg·(L·h)-1逐渐上升至15.17 mg·(L·h)-1,随着搅拌强度的增加,NO3--N的转化速率增加变慢. 当搅拌速率由160 r·min-1上升至320 r·min-1(GT值为22~64 s-1)时,硝酸盐转化速率由8.75mg·(L·h)-1提升至14.24mg·(L·h)-1,提升60%,而当搅拌转速从320 r·min-1上升至700 r·min-1 (GT值为89~165 s-1),硝酸盐转化速率提升13%. 也就是说搅拌GT值为22~165 s-1时的变化对硝酸盐转化速率有较大的影响. 而硝酸盐和氨同步转化的比例的变化几乎和硝酸盐转化强度变化一致. 当搅拌GT值为22~64 s-1时,Δn(NH4+)∶Δn(NO3-)由0.43降低至0.38,降低12%,而GT值为89~165 s-1时,Δn(NH4+)∶Δn(NO3-)由0.38降低至0.14,降低63%.
 | 图 4 搅拌强度及pH值环境对耦合脱除硝酸盐和氨反应的影响 Fig. 4 Effects of stirring intensity and pH value on the simultaneous ammonia and nitrate removal reaction |
控制搅拌GT值为22 s-1,pH对耦合反应的影响如图 4(b)所示,当pH值为5.08~8.08时,随着pH值的升高,NO3--N的转化速率由6.72 mg·(L·h)-1增加10.69 mg·(L·h)-1,当pH值为8.91,转化速率急剧降低至6.88 mg·(L·h)-1. 在该pH值范围的变化过程中硝酸盐和氨同步转化的比例的变化也几乎和硝酸盐转化强度变化一致. 当pH值为5.08提高至8.08过程中,Δn(NH4+)∶Δn(NO3-)由0.05提高至0.43. 当pH值提高为8.91时,Δn(NH4+)∶Δn(NO3-)又降低至0.30.
3 讨论 3.1 耦合厌氧氨氧化反应对硫自养反硝化产生硫酸盐的影响由图 5可见,当16 d添加NO3--N为200mg·L-1左右时,反应体系内Δn(SO42-)∶Δn(NO3-)稳定至1.21±0.06,其值与理论值1.1接近[21],当加入60mg·L-1 NH4+-N后,该摩尔比值迅速降低至1.01±0.10. 两个阶段过程中NO3--N损失量为150mg·L-1左右[图 2(a)和图 3(a)],加入60mg·L-1氨氮后,硫自养反硝化脱氮产生的SO42--S平均减少了40mg·L-1,说明自养反硝化耦合厌氧氨氧化反应可有效遏制硫酸盐的产生.

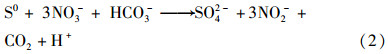



 | 图 5 硫自养反硝化与硫自养反硝化耦合厌氧氨氧化中硫酸盐产率、 IC消耗及pH变化对比 Fig. 5 Comparison of sulfate generation,alkalinity consumption and pH variation between sulfur-based denitrification and simultaneous sulfur denitrification and anaerobic ammonia oxidation |
由于反应器完全密封,脱气口和出水口均做水封处理并且进水已做氮气吹脱除氧,其始终保持厌氧环境,故当添加氨氮后,氨氮只能与硝酸盐还原的亚硝酸盐发生厌氧氨氧化,此时体系内形成硫自养反硝化耦合厌氧氨氧化反应过程. 如图 6所示,其中部分的硝酸盐转化为亚硝酸盐,其再与单质硫反应,最终生成氮气. 另一部分的硝酸盐转化为亚硝酸盐后与氨发生厌氧氨氧化反应. 实测的NH4+-N参与厌氧氨氧化的量为30~50mg·L-1,根据反应式(5)推测可能进行的半自养反硝化的硝酸盐量为39~66mg·L-1,占硝酸盐反应量的26%~44%. 依据公式(2)和(3)估算,这部分硝酸盐只被还原为亚硝酸盐,可降低的SO42--S为36~60mg·L-1. 而实测平均降低量为40mg·L-1. 由于ANAMMOX污泥中,存在一定比例的亚硝化菌(<10%),厌氧反应体系进水中残存的溶解氧可氧化部分氨[30, 31],这导致了硫酸盐实测值略低于理论值.
 | 图 6 硫自养反硝化耦合厌氧氨氧化反应机制 Fig. 6 Mechanism of simultaneous sulfur based denitrification and anaerobic oxidation |
16~30 d为硫自养反硝化稳定阶段,其单位硝态氮消耗的IC量为0.72 mg·L-1±0.15mg·L-1,31~59 d为硫自养反硝化耦合厌氧氨氧化阶段,其单位硝态氮消耗的IC量为0.51 mg·L-1±0.11mg·L-1(图 5). 可以看出,添加60mg·L-1氨氮后,单位硝态氮消耗IC的量平均减少了0.21mg·L-1,可知耦合反应体系可以降低IC的消耗量.
当未添加氨氮的情况下,16~30 d,出水pH值平均为6.5,单位硝态氮降低的pH值为0.012±0.003,当添加60mg·L-1氨氮后,出水pH值平均升高为7.2,单位硝态氮降低的pH值为0.007±0.001(图 5),氨氮的添加也可以减少pH的变化程度.
 | 图 7 搅拌强度对反应硫粒及污泥的影响 Fig. 7 Effect of stirring intensity on sulfur granule and sludge |
本反应体系中pH的变化由氢离子产生、 氢离子消耗、 缓冲体系强弱决定. 硫自养反硝化过程消耗IC而产生氢离子[26],厌氧氨氧化过程消耗氢离子但几乎不消耗IC[29]. 由此认为,当耦合了厌氧氨氧化过程,部分硫自养反硝化反应只将硝酸盐还原至亚硝酸盐,其减少了部分IC的消耗和pH的产生,同时厌氧氨氧化反应产生了部分的氢离子. 耦合反应体系较单纯的硫自养反硝化反应体系,其IC消耗减少,氢离子消耗减少,故体系碱度消耗及环境pH的变化都有所减缓.
3.3 硫自养反硝化耦合厌氧氨氧化反应的最佳控制条件由图 4(a)可见,由于固-液反应体系的特殊性质,搅拌强度对硝酸盐的转化和耦合脱氨有较大的影响,当搅拌强度GT为22~64 s-1时,Δn(NH4+)∶Δn(NO3-)减少不明显(由0.43降低至0.38,降低12%),硝酸盐转化速率增量较大[由8.75mg·(L·h)-1提升至14.24mg·(L·h)-1,提升60%,无氨对照组与其一致,结果未列出]. 搅拌强度引起了传质速率的改变. 本研究发现在传统的亚硝酸盐型厌氧氨氧化体系中(微生物-液体传质),ANAMMOX颗粒污泥在搅拌强度GT大于64 s-1后,反应速率没有明显的改变,说明了在本反应体系中ANAMMOX 反应不需要考虑搅拌强度的影响. 而硫自养反硝化反应(固体-微生物-液体传质),显然固相传递限制要高于液相传质限制,故反应体系中以固相传质的限制作为主要探讨因素.
一方面,硝酸盐与单质硫反应需要一定的传质速率,当搅拌强度GT值从22 s-1提高至64 s-1时,硝酸盐转化速率增加60%,表明该过程中传质速率为限制性因素之一. 而当搅拌强度GT继续提高至165 s-1过程中,硝酸盐转化速率仅增加13%,说明传质速率并不是限制性因素. 另一方面,硝酸盐与单质硫反应的中间产物亚硝酸盐可以通过强化搅拌使其更多地从固-液界面传递到液相,从而为厌氧氨氧化反应提供有力底物支撑,但过大的搅拌强度又会使得单质硫固体颗粒解体(如图 7). 当搅拌强度在160~320 r·min-1时,Δn(NH4+)∶Δn(NO3-)为0.43~0.38,硫粒未出现较大的破碎,说明此时的搅拌强度释放了较多的亚硝酸盐,厌氧氨氧化微生物在对亚硝酸盐的底物竞争中处于优势地位. 当搅拌GT值在22~64 s-1时,Δn(NH4+)∶Δn(NO3-)快速下降至0.14,硫粒已经破碎成较小颗粒. 此时虽然搅拌强度加大增加了亚硝酸与液相交换的机会,但硫颗粒破碎也增加了硫的接触面积,当厌氧氨氧化微生物与硫自养反硝化微生物竞争亚硝酸盐时,硫自养反硝化处于主体地位,故Δn(NH4+)∶Δn(NO3-)较低.
pH值是影响微生物代谢的重要因素之一,在硫自养反硝化耦合厌氧氨氧化反应体系内存在着两种微生物的竞争,pH值调节两类微生物的活性以达到同步脱除硝酸盐和氨的目的. 有报道认为硫自养反硝化的最适pH值为6.8~7.2[8~10],而厌氧氨氧化的最适pH值为7.5~8.1[32]. 如图 4(b)所示,硫自养反硝化和厌氧氨氧化反应活性几乎是同步的,当pH值为8.08时,Δn(NH4+)∶Δn(NO3-)达到最大值0.43,NO3--N的转化速率最大值10.69 mg·(L·h)-1,说明在该反应体系下,一方面是由于进水较高的硝酸盐浓度和较低的氨浓度,另一方面ANAMMOX反应消耗单位氨升高的pH值少(单纯的消耗氢离子),而硫自养反硝化消耗单位硝酸盐降低的pH值多(消耗IC的过程). 这两个原因导致了耦合反应需要较高的pH值环境.
4 结论(1)全混式厌氧搅拌罐接种厌氧氨氧化污泥(湿重)100g·L-1,反应器启动16~30 d,硝态氮脱除负荷达0.56~0.71 kg·(m3·d)-1,接种ANAMMOX污泥可快速启动单质硫自养反硝化反应器.
(2)接种厌氧氨氧化污泥启动的硫自养反硝化反应体系内添加60mg·L-1氨氮,可实现部分硫自养反硝化耦合厌氧氨氧化,反应体系内Δn(SO42-)∶Δn(NO3-)由1.21±0.06降低至1.01±0.10,ΔIC∶ΔNO3--N由0.72±0.1降低至0.51±0.11,出水pH值由6.5上升至7.2. 单位硝酸盐产生的硫酸盐有效减少,单位硝酸盐消耗的碱度显著降低.
(3)硫自养反硝化耦合厌氧氨氧化反应体系连续运行29 d,出水氨氮稳定在10.1~19.2mg·L-1,硝酸盐去除负荷稳定在0.66~0.88 kg·(m3·d)-1,说明该硫自养反硝化与厌氧氨氧化可长期共存于同一反应器内.
(4)在搅拌GT值为22~64 s-1,pH值为8.08时,耦合反应Δn(NH4+)∶Δn(NO3-)最高达到0.43,硝酸盐转化速率提升60%.
| [1] | Ali M, Okabe S. Anammox-based technologies for nitrogen removal: advances in process start-up and remaining issues[J]. Chemosphere, 2015, 141: 144-153. |
| [2] | Van Hulle S W H, Vandeweyer H J P, Meesschaert B D, et al. Engineering aspects and practical application of autotrophic nitrogen removal from nitrogen rich streams[J]. Chemical Engineering Journal, 2010, 162(1): 1-20. |
| [3] | Abma W R, Schultz C E, Mulder J W, et al. Full-scale granular sludge Anammox process[J]. Biofilm Systems VI, 2007, 55(8-9): 27-33. |
| [4] | Van der Star W R L, Abma W R, Blommers D, et al. Startup of reactors for anoxic ammonium oxidation: experiences from the first full-scale anammox reactor in Rotterdam[J]. Water Research, 2007, 41(18): 4149-4163. |
| [5] | Till B A, Weathers L J, Alvarez P J J. Fe (0)-supported autotrophic denitrification[J]. Environmental Science & Technology, 1998, 32(5): 634-639. |
| [6] | Xia S Q, Zhang Y H, Zhong F H. A continuous stirred hydrogen-based polyvinyl chloride membrane biofilm reactor for the treatment of nitrate contaminated drinking water[J]. Bioresource Technology, 2009, 100(24): 6223-6228. |
| [7] | Sahinkaya E, Yurtsever A, Aktaş Ö, et al. Sulfur-based autotrophic denitrification of drinking water using a membrane bioreactor[J]. Chemical Engineering Journal, 2015, 268(15): 180-186. |
| [8] | Sahinkaya E, Dursun N. Use of elemental sulfur and thiosulfate as electron sources for water denitrification[J]. Bioprocess and Biosystems Engineering, 2015, 38(3): 531-541. |
| [9] | Wan D J, Liu H J, Qu J H, et al. Using the combined bioelectrochemical and sulfur autotrophic denitrification system for groundwater denitrification[J]. Bioresource Technology, 2009, 100(1): 142-148. |
| [10] | Han G B, Park J. NO3--N removal with sulfur-lime porous ceramic carrier (SLPC) in the packed-bed bioreactors by autosulfurotrophic denitrification[J]. Journal of the Taiwan Institute of Chemical Engineers, 2012, 43(4): 591-596. |
| [11] | Flere J M, Zhang T C. Nitrate removal with sulfur-limestone autotrophic denitrification processes[J]. Journal of Environmental Engineering, 1999, 125(8): 721-729. |
| [12] | Liu L H, Koenig A. Use of limestone for pH control in autotrophic denitrification: batch experiments[J]. Process Biochemistry, 2002, 37(8): 885-893. |
| [13] | Wan D J, Liu H J, Liu R P, et al. Study of a combined sulfur autotrophic with proton-exchange membrane electrodialytic denitrification technology: Sulfate control and pH balance[J]. Bioresource Technology, 2011, 102(23): 10803-10809. |
| [14] | Lee D U, Lee I S, Choi Y D, et al. Effects of external carbon source and empty bed contact time on simultaneous heterotrophic and sulfur-utilizing autotrophic denitrification[J]. Process Biochemistry, 2001, 36(12): 1215-1224. |
| [15] | Oh S E, Yoo Y B, Young J C, et al. Effect of organics on sulfur-utilizing autotrophic denitrification under mixotrophic conditions[J]. Journal of Biotechnology, 2001, 92(1): 1-8. |
| [16] | Liu H J, Jiang W, Wan D J, et al. Study of a combined heterotrophic and sulfur autotrophic denitrification technology for removal of nitrate in water[J]. Journal of Hazardous Materials, 2009, 169(1-3): 23-28. |
| [17] | Wang H Y,Qu J H. Combined bioelectrochemical and sulfur autotrophic denitrification for drinking water treatment[J]. Water Research, 2003, 37(15): 3767-3775. |
| [18] | Sahinkaya E, Dursun N, Kilic A, et al. Simultaneous heterotrophic and sulfur-oxidizing autotrophic denitrification process for drinking water treatment: control of sulfate production[J]. Water Research, 2011, 45(20): 6661-6667. |
| [19] | Zhang T C, Lampe D G. Sulfur: limestone autotrophic denitrification processes for treatment of nitrate-contaminated water: batch experiments[J]. Water Research, 1999, 33(3): 599-608. |
| [20] | 国家环境保护总局. 水和废水监测分析方法[M]. (第四版). 北京: 中国环境科学出版社, 2002. 258-282. |
| [21] | Sahinkaya E, Dursun N. Sulfur-oxidizing autotrophic and mixotrophic denitrification processes for drinking water treatment: elimination of excess sulfate production and alkalinity requirement[J]. Chemosphere, 2012, 89(2): 144-149. |
| [22] | Soares M I M. Denitrification of groundwater with elemental sulfur[J]. Water Research, 2002, 36(5): 1392-1395. |
| [23] | Moon H S, Shin D Y, Nam K, et al. A long-term performance test on an autotrophic denitrification column for application as a permeable reactive barrier[J]. Chemosphere, 2008, 73(5): 723-728. |
| [24] | Mora G, de Giner G F, Andara G, et al. Effect of organic carbon shock loading on endogenous denitrification in sequential batch reactors[J]. Bioresource Technology, 2003, 88(3): 215-219. |
| [25] | Chen G H, Ozaki H, Terashima Y. Endogenous denitrification in biofilm[J]. Water Science and Technology, 1992, 26(3-4): 523-534. |
| [26] | Koenig A, Liu L H. Kinetic model of autotrophic denitrification in sulphur packed-bed reactors[J]. Water Research, 2001, 35(8): 1969-1978. |
| [27] | Batchelor B, Lawrence A W. A kinetic model for autotrophic denitrification using elemental sulfur[J]. Water Research, 1978, 12(12): 1075-1084. |
| [28] | Kuypers M M, Sliekers A O, Lavik G, et al. Anaerobic ammonium oxidation by anammox bacteria in the Black Sea[J]. Nature, 2003, 422(6932): 608-611. |
| [29] | Strous M, Heijnen J J, Kuenen J G, et al. The sequencing batch reactor as a powerful tool for the study of slowly growing anaerobic ammonium-oxidizing microorganisms[J]. Applied Microbiology and Biotechnology, 1998, 50(5): 589-596. |
| [30] | Liang Y H, Li D, Zhang X J, et al. Performance and influence factors of completely autotrophic nitrogen removal over nitrite (CANON) process in a biofilter packed with volcanic rocks[J]. Environmental Technology, 2015, 36(8): 946-952. |
| [31] | Liang Y H, Li D, Zhang X J, et al. Stability and nitrite-oxidizing bacteria community structure in different high-rate CANON reactors[J]. Bioresource Technology, 2015, 175: 189-194. |
| [32] | Jaroszynski L W, Cicek N, Sparling R, et al. Importance of the operating pH in maintaining the stability of anoxic ammonium oxidation (ANAMMOX) activity in moving bed biofilm reactors[J]. Bioresource Technology, 2011, 102(14): 7051-7056. |