 2015, Vol. 36
2015, Vol. 36



砷是一种有毒的非金属元素,在环境中广泛存在[1,2]. 一般而言,环境中的砷主要以As(Ⅴ)和As(Ⅲ)为主[3],二者因环境条件改变或在环境微生物作用下可以相互转化,在厌氧环境中砷主要以As(Ⅲ)形态为主[4,5]. 大量研究[6, 7, 8, 9, 10]证实厌氧微生物作用所引起的硫酸盐还原过程强烈影响砷的地球化学行为,其主要原因是硫酸盐还原产生的S(Ⅱ)离子可以与As(Ⅲ)形成砷的硫化物矿物,如雌黄、 雄黄、 毒砂等,因此砷的环境归趋路径中,形成硫化砷成为砷形态转化中很重要的一个过程. 同时在一些砷污染的天然湖泊、 地热和温泉等环境中硫化物含量也很高[11],一般研究认为砷的硫化物是比较稳定的矿物,有研究采用添加硫酸盐的方法修复水体砷污染也取得了一定的成效[12]. 但目前也有研究发现硫化条件下硫化砷不是很稳定的,过硫化条件下可能发生硫化砷的溶解[13]. 因此针对硫化砷在环境中的形态与稳定性的研究对于地下水砷污染修复具有重要的意义,特别是理解高硫的环境中,如热泉环境中砷的转化过程与机制也是重要的补充.
雌黄矿物中砷的形态和稳定性与环境条件相关. 在16~18℃条件下雌黄在纯水中的溶解度为2.1×10-3 mmol ·L-1[14]. pH值是影响雌黄溶解度的重要参数,Webster[15]研究了雌黄溶解于水溶液的平衡常数以及形态变化的化学方程式. 在25℃和90℃,pH < 7的稀溶液中,H3AsO03为雌黄溶解后形成砷的主要存在形态,在这一条件下,当介质中加入硫离子时,会与溶解态砷重新形成雌黄,发生再沉淀现象. 随着pH的变化,雌黄的溶解度发生改变. 当环境由中性变为碱性条件,雌黄的溶解性逐渐增加,当溶液从中性pH 7到碱性pH 12,雌黄溶出砷浓度从0.82mmol ·L-1增加到3.47 mmol ·L-1[13]. 雌黄溶解后产物的形态也与环境条件相关,有研究认为在pH 2~8时,亚砷酸根是雌黄溶解后的水溶液中主要存在的形态,约占总砷形态的70%~90%[16]. 这些研究主要关注温度、 pH的影响,对雌黄溶解度及溶解产物的研究方面也都是基于有氧条件下的研究结果. 雌黄中与砷结合的硫离子是非常容易氧化的元素,游离硫离子的量直接影响砷的形态[17]. 因此厌氧条件下雌黄的溶解过程与有氧条件下会有很大区别,但目前针对雌黄的稳定性方面的研究较多集中在冶金的尾矿中硫化砷的风化溶解方面[13,18],对于厌氧条件下硫化砷的溶解过程及产物组成的研究非常缺乏.
为了考察自然环境条件下雌黄的溶解产物组成及其赋存形态变化,本研究利用X射线吸收光谱法(XAS)和数据拟合方法分析雌黄溶解产物在厌氧中性条件下有硫和无硫两种介质中的产物形态,并借助EXAFS分析产物中AsⅢ—S和AsⅢ—O的键长和可能的产物组成比例. 研究雌黄在厌氧条件下的形态,以期为天然水体与沉积环境中砷的形态变化与污染修复的建立提供理论补充,同时也有助于进一步完善砷的地球化学循环模型. 1 材料与方法 1.1 雌黄矿物和溶液配制
雌黄来源于天然矿物,经过破碎研磨,制备成为较均匀的雌黄粉末(85% < 100 μm),之后未做任何预处理. 实验所用到的液体溶液都是用厌氧水配制的. 1.2 雌黄的溶解实验
称取0.02 g雌黄粉末于厌氧瓶中,在厌氧箱中进行溶解实验. 无硫条件下的研究利用5mol ·L-1氢氧化钠溶液进行; 利用氢氧化钠溶液吸收硫化氢气体获得的纯硫化钠溶液(56mmol ·L-1)进行有硫条件下的研究. 使用一定浓度的氢氧化钠和盐酸调节溶液pH值约为7,并保持恒定. 原子荧光光谱法[19,20]测定两种样品溶液中的总砷浓度约为(16.3±0.2)mmol ·L-1. 反应平衡120 h后研究不同环境条件下的形态. 1.3 XAS法测定溶解样品
在厌氧箱中取溶解120 h的液体溶液,针管过滤器过滤获得透明溶液,使用小自封袋封装,用思高胶带密封隔绝氧气,密封于厌氧瓶中. 砷的k边EXAFS(拓展X射线吸收)光谱测试,使用北京同步辐射实验室1W1B光束(XAFS站)进行. 实验方法参见文献[21].
XANES表征实验中,选择亚砷酸钠、 砷酸钠、 雄黄和雌黄作为测试As的标准物. 其中雄黄和雌黄均为天然矿物,亚砷酸钠和砷酸钠为分析纯试剂. 这些标准物经过研磨制备为砷的标准物. 2 结果与分析 2.1 有硫和无硫条件下雌黄溶解溶液中的As同步辐射结果
不同标准样品的XANES数据对比表明不同形态的砷中砷原子k边X射线吸收谱的吸收边位置有明显的差异,而砷的氧化态的变化可以导致砷吸收能量的位置变化,因此与标准物中砷的吸收边能量位置比较,可以根据As的XANES吸收边位置变化和R空间变化确定砷的价态和价键的变化. 在以As2S3代表AsⅢ—S标准,AsⅢ—S的吸收峰位置是11871.8 eV,以NaAsO2代表AsⅢ—O的标准,AsⅢ—O键的吸收峰位置是11873.6 eV. 本研究结果见图 1,在有硫体系下平衡溶液中砷的能量峰值与雌黄固体标准物接近,处于11871.9 eV,说明雌黄溶解产物在pH 7条件下可能是以AsⅢ—S为主的赋存形态. 而在无硫条件下的溶液中测得的砷的吸收峰峰形变宽,峰值为11872.8 eV,处于AsⅢ—S和AsⅢ—O之间,说明溶液中的硫代砷形态可能为AsⅢ—S和AsⅢ—O的混合物,也就是混合产物中存在砷氧键和砷硫键的共同作用的结果. 由于所有产物的吸收峰的能量位置都与三价砷标准的吸收峰更接近,因此可以确定实验中没有发生氧化现象,即没有五价砷的形态存在.
 | 图 1 砷的XANES光谱图谱 Fig. 1 Arsenic XANES spectra |
为了更准确地确定溶液中的砷与硫结合产生的形态,还需要借助其他方法对数据进行分析处理,结合R空间和k空间的相关数据对有硫和无硫条件下雌黄溶解溶液中的砷形态进行形态分析,获得更准确的信息以预测砷形态的化学结构. 本研究使用XANES线性组合拟合方法,对有硫和无硫体系中所得各种砷形态的砷进行数据拟合,获得结果见表 1. 溶解产物在有硫的中性条件下主要是硫代三价砷(约88.2%)形态存在,而亚砷酸钠形态的比例很低,大约为11.8%. 而对应的无硫条件下溶解产物不仅含有硫代三价砷,约为总砷形态的43.7%,同时溶解产物中也含有较高比例的游离态亚砷酸钠形态约占56.3%,样品中没有拟合出硫代五价砷形态. 因此,厌氧条件下在pH 7溶液中雌黄溶解形成产物的硫代三价砷可能是主要的砷存在形态.
| 表 1 XANES线性组合拟合样品中的形态 Table 1 Sample speciation based on XANES Linear Combination of Fits (LCF) |
雌黄是由As—S一种化学键组成的链式结构,当雌黄溶解在有硫和无硫的介质中,可以产生不同的砷硫形态产物. 根据不同产物中As—O和As—S价键长度的差异性,可以作为砷形态鉴定的依据. 如亚砷酸根中的三价砷与周围的氧结合形成的空间构型为三角锥形,而砷酸根中的砷是与 周围的4个氧结合形成的空间构型为四棱锥,因 此两者之间的价键长度就存在不同[22]. 本研究利用同步辐射EXAFS检测了有硫和无硫条件下雌黄的溶解产物.
本研究中测定了以硫化钠为介质代表有硫体系,以氢氧化钠溶液作为溶解介质的无硫体系的雌黄产物的扩展X射线吸收光谱(EXAFS),经过Artemis软件[23]进行拟合分析后得出在中性厌氧环境条件下溶解产物中砷氧键和砷硫键的长度,拟合结果见图 2.
 | 圆圈是EXAFS数据,黑色的线是拟合结果. 雌黄、 亚砷酸钠和砷酸钠为标准 图 2 R空间谱图拟合和As-k3光谱拟合结果 Fig. 2 Arsenic corresponding Fourier transforms (FT) and k3-weighted EXAFS functions of samples and references |
在标准物质雌黄和亚砷酸钠中,AsⅢ—O键的键长一般为0.178 nm,AsⅢ—S键的键长为0.224~0.234 nm,而五价砷的AsV—O和AsV—S键的键长分别为0.170 nm和0.213~0.218 nm[24]. 本研究中EXAFS数据拟合结果表明,有硫条件下溶解产物的As—S键和无硫条件下溶解产物As—S键都与雌黄矿物标准中的AsⅢ—S键长接近,分别为0.223 nm和0.222 nm,而无硫条件下溶解产物中As—O键的键长接近亚砷酸盐标准物中AsⅢ—O键长的长度(为0.178 nm),见表 2. 因此有硫条件下溶解产物主要含有As—S的峰,可能为全硫代的硫代砷形态,而无硫条件下溶解产物中主要为含有As—S和As—O的峰,再结合能量峰位置分析,说明溶液中砷的结合形态可能为硫代亚砷酸盐(AsⅢ—S)和亚砷酸盐(AsⅢ—O)的混合物,即部分硫代的亚砷酸根.
| 表 2 实验样品的EAXFS拟合结果 Table 2 EXAFS fitting results for experimental samples |
雌黄是环境中常见的含砷矿物,其溶解过程受环境的温度、 pH和环境介质离子,特别是硫离子含量的影响[13,15]. 本文针对中性pH条件下雌黄溶解产物在有硫和无硫两种介质中具体砷的形态进行对比研究,结合已有报道发现硫砷比不同,所得产物结构不同. 本研究中,雌黄硫添加体系中的溶解过程硫代砷的产生过程主要发生如下反应[25]:
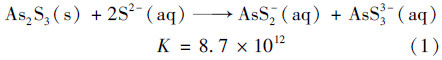
由方程式可见,有硫条件下雌黄溶解形成的硫代砷砷形态主要为两种全硫代的三价砷:AsS2-和AsS3-3. Webster[15]研究雌黄溶解产物也发现在硫化钠体系中可以存在几种硫代砷化合物:AsS2-、 HAs2S-4、 H2As3S-6,都是属于全硫代的亚砷酸盐. 这与本实验得出的结论相一致.对于无硫体系而言,式(2)[25]中给出雌黄溶解在氢氧化钠中产生的形态为混合硫代的亚砷酸盐,其形态分别为AsS2-和AsS(OH)2. Webster的研究中没有发现硫代亚砷酸盐[15],只有亚砷酸盐.
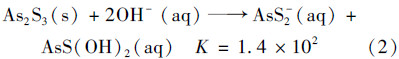
同时,在本研究中发现,无论是在有硫还是无硫添加条件下,体系中都会产生少量的亚砷酸钠. Beak等[19]的研究中也发现了亚砷酸和硫离子反应可以形成硫代亚砷形态和少量的亚砷酸盐. 可能原因是由pH值和溶液中含有的配位体种类对于溶液中砷形态的影响造成的. 即在中性条件下,由于溶液中与SH-具有竞争性的氢氧根配位体含量较多,会阻滞硫代反应的发生,雌黄溶解形成的砷硫阴离子形态中的硫可能会被溶液中的OH-取代,形成砷氧阴离子,即亚砷酸根的形式[26]. 而且产物中亚砷酸盐发生硫代的比例与硫砷比相关. 所以,相比于有硫条件下雌黄的溶解反应,无硫条件下的溶解产物中可以形成较高含量的亚砷酸钠,而且在中性pH值下,也更有利于亚砷酸根的形成. 总之,雌黄溶解产物中砷形态与反应体系的pH有关,反应过程是否严格避氧也是影响产物的主要因素之一[13]. 因此,无论是有硫溶解体系还是无硫溶解,雌黄溶解产物中砷形态是以硫代三价砷形态和部分的亚砷酸根形态为主.
关于雌黄溶解产物的研究中,目前提出的硫代砷产生机制[13]是基于负二价硫离子在溶液中与三价砷形成硫代砷形态的解释. 对于雌黄碱性溶解过程中,雌黄表面可能会先释放出亚砷酸根和负二价硫离子,之后两者发生硫代反应,结合形成硫代三价砷形态. 而在有氧条件下产生硫代三价砷进而被氧化为硫代五价砷形态. 此外,早期的研究也发现游离的砷离子和硫离子结合可以形成硫代砷的形态[27]. 对于不同体系中硫代砷产生机制的研究需要继续深入,以获得数据支持. 4 结论
(1)在砷的XANES谱图中,pH中性条件下,雌黄在有硫和无硫条件下的溶解产物中砷形态的吸收能量峰值在雌黄和亚砷酸钠之间,说明形成硫代砷和亚砷酸盐的混合物.
(2)过量的硫离子有利于硫代亚砷酸盐的形成,线性拟合分析确定在有硫体系中硫代亚砷酸盐形态占总砷形态的88.2%,亚砷酸盐形态占11.8%,而在无硫体系中,硫代三价砷形态占总砷形态的43.7%,亚砷酸盐形态占56.3%.
(3)利用EXAFS拟合分析进一步获得两种体系中雌黄溶解形成的砷硫键和砷氧键的信息,在有硫体系中硫代砷是以全硫代为主,而无硫体系中硫代亚砷酸盐是部分硫代为主.
致谢: 本研究样品检测得到北京同步辐射实验室的技术支持,以及站上各位老师的帮助,在此表示感谢.
| [1] | Matschullat J. Arsenic in the geosphere-a review[J]. Science of the Total Environment, 2000, 249 (1-3): 297-312. |
| [2] | 陶俊, 张仁健, 段菁春, 等. 北京城区PM2.5中致癌重金属季节变化特征及其来源分析[J]. 环境科学, 2014, 35 (2): 411-417. |
| [3] | 李磊, 任景玲, 刘素美, 等. 桑沟湾溶解态无机砷的分布、季节变化及影响因素[J]. 环境科学, 2014, 35 (7): 2705-2713. |
| [4] | Islam F S, Gault A G, Boothman C, et al. Role of metal-reducing bacteria in arsenic release from Bengal delta sediments[J]. Nature, 2004, 430 (6995): 68-71. |
| [5] | 杨会, 谢先军, 段萌语. 不同厌氧环境下土著微生物对沉积物砷释放的影响[J]. 环境科学与技术, 2014, 37 (6): 44-48. |
| [6] | Stucker V K, Williams K H, Robbins M J, et al. Arsenic geochemistry in a biostimulated aquifer: an aqueous speciation study[J]. Environmental Toxicology and Chemistry, 2013, 32 (6): 1216-1223. |
| [7] | Stucker V K, Silverman D R, Williams K H, et al. Thioarsenic species associated with increased arsenic release during biostimulated subsurface sulfate reduction[J]. Environmental Science & Technology, 2014, 48 (22): 13367-13375. |
| [8] | Ahmann D, Krumholz L R, Hemond H F, et al. Microbial mobilization of Arsenic from sediments of the Aberjona Watershed[J]. Environmental Science & Technology, 1997, 31 (10): 2923-2930. |
| [9] | Zhang J Y, Kim H, Townsend T. Methodology for assessing thioarsenic formation potential in sulfidic landfill environments[J]. Chemosphere, 2014, 107: 311-318. |
| [10] | Stauder S, Raue B, Sacher F. Thioarsenates in sulfidic waters[J]. Environmental Science & Technology, 2005, 39 (16): 5933-5939. |
| [11] | Planer-Friedrich B, Fisher J C, Hollibaugh J T, et al. Oxidative transformation of trithioarsenate along alkaline geothermal drainages-abiotic versus microbially mediated processes[J]. Geomicrobiology Journal, 2009, 26 (5): 339-350. |
| [12] | O'Day P A. Chemistry and mineralogy of arsenic[J]. Elements, 2006, 2 (2): 77-83. |
| [13] | Suess E, Planer-Friedrich B. Thioarsenate formation upon dissolution of orpiment and arsenopyrite[J]. Chemosphere, 2012, 89 (11): 1390-1398. |
| [14] | Biltz W. Einige Versuche über ultra-mikroskopische Loslichkeitsbestimmung (in German)[J]. Zeitschrift für Physikalische Chemie, 1907, 58: 288-292. |
| [15] | Webster J G. The solubility of A2S3 and speciation of As in dilute and sulphide-bearing fluids at 25 and 90℃[J]. Geochimica et Cosmochimica Acta, 1990, 54 (4): 1009-1017. |
| [16] | Floroiu R M, Davis A P, Torrents A. Kinetics and mechanism of As2S3(am) dissolution under N2[J]. Environmental Science & Technology, 2004, 38 (4): 1031-1037. |
| [17] | Bostick B C, Fendorf S. Arsenite sorption on troilite (FeS) and pyrite (FeS2)[J]. Geochimica et Cosmochimica Acta, 2003, 67 (5): 909-921. |
| [18] | Haffert L, Craw D. Mineralogical controls on environmental mobility of arsenic from historic mine processing residues, New Zealand[J]. Applied Geochemistry, 2008, 23 (6): 1467-1483. |
| [19] | Beak D G, Wilkin R T, Ford R G, et al. Examination of arsenic speciation in sulfidic solutions using X-ray absorption spectroscopy[J]. Environmental Science & Technology, 2008, 42 (5): 1643-1650. |
| [20] | Couture R M, Rose J, Kumar N, et al. Sorption of arsenite, arsenate, and thioarsenates to iron oxides and iron sulfides: a kinetic and spectroscopic investigation[J]. Environmental Science & Technology, 2013, 47 (11): 5652-5659. |
| [21] | Xu L Y, Zhao Z X, Wang S F, et al. Transformation of arsenic in offshore sediment under the impact of anaerobic microbial activities[J]. Water Research, 2011, 45 (20): 6781-6788. |
| [22] | Tossell J A, Zimmermann M D. Calculation of the structures, stabilities, and vibrational spectra of arsenites, thioarsenites and thioarsenates in aqueous solution[J]. Geochimica et Cosmochimica Acta, 2008, 72 (21): 5232-5242. |
| [23] | Ravel B, Newville M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT[J]. Journal of Synchrotron Radiation, 2005, 12: 537-541. |
| [24] | Suess E, Scheinost A C, Bostick B C, et al. Discrimination of thioarsenites and thioarsenates by X-ray absorption spectroscopy[J]. Analytical Chemistry, 2009, 81 (20): 8318-8326. |
| [25] | Li W. Synthesis and solubility of arsenic tri-sulfide and sodium arsenic oxy-sulfide complexes in alkaline sulfide solutions[D]. Hebei: Hebei University of Technology, 2013. 30-31. |
| [26] | Rochette E A, Bostick B C, Li G C, et al. Kinetics of arsenate reduction by dissolved sulfide[J]. Environment Science & Technology, 2000, 34 (22): 4714-4720. |
| [27] | Wood S A, Tait C D, Janecky D R. A Raman spectroscopic study of arsenite and thioarsenite species in aqueous solution at 25℃[J]. Geochemical Transactions, 2002, 3 (4): 31-39. |