 2015, Vol. 36
2015, Vol. 36



2. 中国科学院生态环境研究中心环境生物技术重点实验室, 北京 100085
2. Key Laboratory of Environmental Biotechnology, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing 100085, China
氯代硝基芳香类抗生素氯霉素(chloramphenicol,CAP),其作为有效的广谱抗生素在世界范围内已经广泛用于处理许多种致病菌,然而由于氯霉素可以引起潜在的致癌性和遗传毒性[1,2],美国食品与药品管理局(USFPA)已经禁止在食品生产的动物上使用氯霉素. 然而在许多国家仍然被广泛使用,在这些国家的污水处理厂的进水和出水中都可以检测出低浓度的氯霉素残留[3, 4, 5, 6],例如在香港地区污水处理厂中出水中检测氯霉素为3~1 050 ng ·L-1[7],在北京地区的养猪场废水中为1.49~11.5 μg ·L-1[8],在贵阳的城市污水中甚至可以高达5.8~47.4 μg ·L-1[9]. 更令人担忧的是在全世界范围内,在不同的污水处理厂,饮用水处理厂中,均可以频繁检测到氯霉素抗性基因和其它抗生素抗性基因[10,11],这些由于抗生素残留引起的环境污染问题已经引起全球的广泛关注. 因此在废水处理过程中,去除氯霉素的抗细菌活性和进一步降低环境生态系统氯霉素抗性细菌和抗性基因显得十分重要.
对于降解氯霉素,主要的方法包括焚烧法[12]、 光催化降解、 微波辐射、 Fenton 氧化技术和电化学氧化技术等物理化学方法,这些方法虽然降解效率较高,但受限于具有一定的能源浪费以及二次污染等缺点. 生物电化学系统(BES)是一种处理效率高且低成本的新技术. BES生物阴极还原降解难降解污染物一直是近年来研究的热点,如生物阴极还原降解硝基芳香烃类污染物(硝基苯及氯霉素)[13, 14, 15, 16, 17]、 偶氮类染料(酸性橙,茜素黄和酸性黑)[18,19]和氯代芳香烃等[20]. 生物阴极的形成,之前的研究主要是以预富集好具有降解特定污染物能力的富集液,之后在阴极给电子环境作用下对这些特定微生物实现嗜电极微生物的阴极挂膜,从而保证阴极微生物的催化降解污染物的特性. 生物阳极具有最重要的特性就是微生物燃料电池(MFC)中提到的理论即: 阳极微生物具有氧化有机物产生电子并通过电极表面将电子传导出去的能力,从而实现了化学能到电能的转换,也是近年来研究的热点问题之一.
生物阳极对抗生素类污染物进行降解报道较少[21,22],尤其是面对氯霉素这类对微生物毒性比较大的抗生素而言,在阳极进行还原降解难度更大,而且对产电微生物的产电能力也有极大的影响,进而导致反应器的运行效能降低. 之前一些学者反转生物阳极进行了生物阴极催化合成氢气和甲烷以及还原硝酸盐研究[23, 24, 25],然而直接反转生物阳极为生物阴极降解有机污染物未见报道. 本研究分析了在长期氯霉素毒性刺激条件下生物阳极在正常产电的同时还原降解氯霉素的可能性,以及生物阳极反转为生物阴极后能否加速还原降解氯霉素.
1 材料与方法 1.1 厌氧接种污泥及培养基配方
阳极微生物挂膜所需微生物菌源来自于哈尔滨太平污水处理厂二沉池污泥,氯霉素(CAP)购买于美国Sigma-Aldrich 公司,其余化学品均为分析纯. 生物阳极液或反转后生物阴极液配方: Na2HPO4 ·12H2O: 11.55 g; NaH2PO4 ·2H2O: 2.77 g; KCl: 0.13 g; NH4Cl: 0.31 g; 乙酸钠: 1.68 g; 矿质元素液1 mL; 微量元素液1 mL; 1 L 去离子水. 非生物阴极液和恒电位仪控制下对电极溶液均为50 mmol ·L-1 磷酸盐缓冲液(PBS),序批式降解实验均添加35 mg ·L-1氯霉素.
1.2 反应器结构双极室BES反应器为两个聚碳酸酯反应器模块(7 cm×7 cm×4 cm)组装而成,每个模块具有中空呈圆柱形的腔体(直径5 cm,长4 cm). 模块用螺栓连接,左右各用一块聚碳酸酯平板(7 cm×7 cm×1 cm)密封. 中间用阳离子交换膜(Ultrex CMI-7000,膜国际,美国)分隔,单个极室体积为85 mL. 阴阳极均为碳刷(直径4 cm,长3 cm,TOHO TENAX Co.,Ltd.,日本). 碳刷是用两根钛丝(直径 1 mm,宝鸡力兴钛业,中国)旋钮碳纤维而制成. 阳极生物膜的降解氯霉素驯化实验采用无隔膜升流式生物电催化反应器(UBER)完成[14]. 二者反应器均外接10 Ω外电阻及0.5 V外加电压(直流电源IT6921,Itech Co. Ltd.,美国). 饱和甘汞参比电极(0.247 V vs. SHE,217 型,上海雷磁,中国)嵌入阴极室用于测量阴极电位. 阴阳极电位及电流通过数据记录仪计数获得(model-2700,吉士利,美国). 本研究中报道的电极电位数据都已换算成对标准氢电极(SHE).
1.3 反应器的运行首先取10 mL二沉池污泥接种于含有阳极液的BES阳极室中,运行3个循环,每次均接种等量污泥,之后不接种污泥更换阳极液运行3个循环,共6个循环. 待阳极电位降至-0.25 V、 反应器电流稳定在0.2~0.4 mA时表明反应器阳极启动成功. 其次将生物阳极碳刷在厌氧条件下转移至UBER阳极进行生物阳极降解氯霉素驯化实验,加入反应器内的氯霉素浓度由小至大依次增加(5、 10、 20、 35、 60、 80 mg ·L-1). 5 mg ·L-1浓度运行6个循环,其余浓度运行2个循环. 再次将驯化好的生物阳极安装在双极室反应器中运行生物阳极与非生物阴极同步还原降解氯霉素实验,运行3个循环. 最后将生物阳极反转为生物阴极,采用恒电位仪(WMPG1000K8,WonATech Co.,韩国)控制阴极电位-0.40 V运行6个循环. 去除生物阴极的生物膜继续运行3个循环作为非生物阴极对照实验.
1.4 分析方法高效液相色谱法(model e2695,沃特斯,美国)分析氯霉素浓度和还原降解产物形成,紫外检测器的检测波长为275 nm 检测氯霉素和氯霉素乙酰化产物(Acetyl); 310 nm 检测亚硝基(NO); 250 nm 检测芳香胺产物(AMCl2、 AMCl和AM). 色谱柱是C18 反向色谱柱(5 μm; 5 mm×250 mm,沃特斯,美国),柱温设定为30℃,流动相配比为55%甲醇/45%水,流速为0.8 mL ·min-1. 高效液相色谱-质谱联用仪(LC-MS)由液相色谱(model-1200,安捷伦,美国)搭配高精度飞行时间四极杆质谱(model-6520,安捷伦,美国)组成,色谱柱采用安捷伦 C18柱 (1.8 μm; 2.1 mm×100 mm),设定负离子模式运行,流动相配比为55%甲醇/45%水,流速为0.10 mL ·min-1,紫外检测器设定波长为250 nm和275 nm. 电化学工作站(CHI-660E,辰华,上海)用于生物阳极反转前后循环伏安(CV)曲线的扫描和交流阻抗(EIS)分析. 本研究中提到的氯霉素还原降解效率(Er,%)通过如下公式获得:
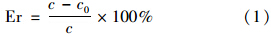
取二沉池污泥接种于含有阳极液的BES反应器阳极,外加0.5 V电压,对电极(非生物阴极)添加35 mg ·L-1氯霉素. 实验结果如图 1(a)所示,在整个420 h的启动阶段,阳极和阴极的电位分别降到-0.15 V和-0.65 V所需的时间约为96 h,在之后5个循环阴阳极电位保持不变,电流则逐渐增加至稳定阶段,表明生物阳极启动成功. 此时的生物阳极表面已经附着大量的产电微生物,产生的电子通过阳极传导到阴极表面进行非生物阴极还原氯霉素. 取出具有产电能力的生物阳极置于无隔膜UBRE反应器进行降解氯霉素的驯化,与双极室反应器的不同之处在于UBER反应器无隔膜且阴阳极同处一室,因此氯霉素的降解除了非生物阴极降解之外还包括了生物阳极潜在的生物降解. 氯霉素的初始驯化浓度为5 mg ·L-1,共运行6个循环. 如图 1(b)所示,当反应器运行至100 h时,阴阳极电位分别维持在0.10 V和0.60 V左右,电位较之前大幅升高说明具有产电能力的阳极产电微生物受到氯霉素的毒性影响. 反应器运行至200 h后,阴阳极电位依次稳定在-0.20 V和-0.70 V左右,而电流值也逐步稳定在0.5~1 mA之间,表明阳极表面产电微生物已经能够在氯霉素存在的条件下进行正常的代谢和产电.
 | 图 1 生物阳极启动及其驯化阶段的电位与电流变化Fig. 1 Changes of potential and current in the process of starting up and acclimation of bioanode |
在5 mg ·L-1浓度下稳定运行后,增加氯霉素浓度进行生物阳极的高浓度驯化实验. 氯霉素浓度从低到高(5~80 mg ·L-1)共6种浓度梯度的降解,分别用准一级动力学方程拟合出降解速率常数k. 如图 2所示,5 mg ·L-1时Er1h为63.81%±3.96%,k值为1.023±0.189,随着氯霉素浓度的增加,Er1h和k值稳步增加,至20 mg ·L-1时Er1h和k值达到最大分别为92.69%±1.81%和2.071±0.222. 表明氯霉素浓度从5 mg ·L-1增加到20 mg ·L-1时,微生物在此浓度范围内没有受到明显的影响. 当氯霉素浓度增加至35 mg ·L-1时,Er1h和k值分别降低至59.45%±3.94%和1.037±0.051,表明生物阳极嗜电极微生物在此浓度下受到较大的影响,其产电能力和降解能力降低. 继续增加氯霉素浓度至60 mg ·L-1时,Er1h和k值又有所回升,继续升高浓度至80 mg ·L-1时,Er1h和k值再次降低. 这种氯霉素降解效率和速率呈阶梯式增加的现象,表明嗜电极微生物与氯霉素浓度存在着抑制与克服的矛盾关系. 该生物阳极对氯霉素的耐受浓度从5 mg ·L-1提升至80 mg ·L-1,表明降解氯霉素的生物阳极驯化成功.
 | 图 2 生物阳极驯化阶段的氯霉素降解效率和速率Fig. 2 Er1h% and k of CAP degradation in the acclimation stage of bioanode |
生物阳极驯化成功后,将其从UBER反应器中取出装入双极室BES反应器,对电极为非生物阴极. 首先生物阳极中不加氯霉素,其对电极非生物阴极单独降解氯霉素为对照实验,研究生物阳极及其对电极非生物阴极中均加入氯霉素条件下阴阳极同步降解氯霉素的特性. 如图 3所示,分别含有35 mg ·L-1氯霉素的生物阳极和非生物阴极的电位分别为-0.19 V和-0.72 V,略高于对照实验的(-0.24 V)和非生物阴极(-0.78 V),而且相对于对照实验电流值较小. 表明含氯霉素的生物阳极在一定程度上受到了氯霉素的影响. 可能由于氯霉素的毒性致使阳极产电微生物产电能力下降或者生物阳极中氯霉素的存在,作为潜在的电子受体竞争了一部分产电微生物产生的电子,导致传导到阴极的电子相对变少,电流变小.
 | 图 3 生物阳极与非生物阴极同步降解氯霉素电位电流变化Fig. 3 Potential and current changes in the simultaneous CAP degradation of the bioanode and abiotic cathode |
生物阳极和非生物阴极同步降解氯霉素过程中,二者还原降解速率常数k分别为0.098 5和0.513 5,而对照实验的k则为0.644 4(图 4). 结果表明生物阳极具有一定的还原降解氯霉素的能力,而此时生物阳极的电位为-0.20 V,此电位下氯霉素很难被电化学还原降解,因此生物阳极氯霉素的降解主要是单纯微生物的降解或者在微生物的作用下降低了还原反应的过电位[14,15],使得氯霉素在相对较高的电位下也有可能发生还原反应. 此外非生物阴极的降解速率常数小于对照实验(0.513 5<0.644 4)的原因是由于添加氯霉素的生物阳极也在一定程度上受到氯霉素毒性的影响,导致生物阳极电位升高,非生物阴极电位也升高,电流变小(图 3),因此氯霉素的还原降解速率变慢. 产物形成方面如图 4所示,添加氯霉素的生物阳极主要有3种还原产物分别为氯霉素的乙酰化产物(Acetyl)、 芳香胺产物(AMCl2)和脱1个Cl的芳香胺产物(AMCl)[14]. 而非生物阴极的产物除了AMCl2和AMCl(主要产物)外还有非生物阴极典型中间产物亚硝基产物(NO)[15],而没有Acetyl. 对照组实验除了以上3种产物之外,还少量生成了脱2个Cl的芳香胺产物(AM),其原因是对照组实验的阴极电位更低,致使AMCl的第二个Cl在较低电位下被还原脱除.
 | 图 4 生物阳极与非生物阴极同步降解氯霉素降解速率及产物形成规律Fig. 4 Degradation rate and products formation rules in the simultaneous CAP degradation of the bioanode and abiotic cathode (a)非生物阴极对照; (b)生物阳极加氯霉素的非生物阴极; (c)生物阳极 |
实验结果如图 5所示,恒定阴极电位为-0.40 V时,生物阴极还原降解氯霉素的速率常数k值为0.263 4,明显高于非生物阴极对照(k=0.160 9),比非生物阴极对照快38%. 表明反转后的生物阴极具有较强的生物催化还原降解氯霉素的能力. 而生物阴极开路实验的降解速率常数k=0.043 5,表明单独的阴极生物膜的生物作用仍然对氯霉素的降解具有一定的作用,但还原降解速率比反转前生物阳极的慢(k=0.098 5),其原因可能是由于阴极电位由原来生物阳极的-0.20 V变为-0.40 V,电位的降低导致微生物活性受到一定影响,降解氯霉素能力有所下降. 从产物形成方面看,生物阴极相对于非生物阴极对照没有积累毒性较大的NO产物,而是少量积累Acetyl后直接生成AMCl2(主要产物)和AMCl,与之前研究结果相似[14,15],并且AMCl2的形成在48 h内仍然具有下降趋势,表明可能继续被还原为其他物质. 而开路实验的产物则大量积累AMCl2和Acetyl的同时,也有AMCl产物,表明单独阴极生物膜对氯霉素也具有脱氯能力.
 | (a) 生物阴极开路对照; (b) 反转后生物阴极; (c) 非生物阴极对照图 5 生物阳极反转生物阴极降解氯霉素速率及产物形成规律Fig. 5 Degradation rate and product formation rules in CAP degradation by biocathode switched from bioanode |
对生物阴极反应体系运行至48 h和144 h时分别取样进行LC-MS分析,结果如图 6(a)所示,48 h时在质谱图中具有明显的负离子核质比291.07的峰被鉴定为AMCl2(具有典型氯同位素簇,291、 293和295). 与图 5(b)中AMCl为主要产物的结果吻合. 如图 6(b),对144 h的样品进行分析发现在质谱图主要出现3个明显的特征峰,负离子模式下核质比分别为293.075 5(具有典型氯同位素簇293、 295和297)、 260.928 1和225.142 1,依次被鉴定为羰基化的AMCl2,羰基还原并脱掉一个Cl的AMCl以及羰基还原并脱掉2个Cl的AM. 表明长时间生物阴极作用下,不仅可以发生硝基还原反应而且可以进行还原脱氯反应,甚至是氯霉素的羰基还原反应,与前面提到的AMCl2在48 h内仍然有下降趋势,可能被还原为其他产物结果相吻合.
 | (a) 48 h生物阴极出水; (b) 144 h生物阴极出水; (c)0 h生物阴极出水对照图 6 生物阴极降解氯霉素产物的分析与鉴定Fig. 6 LC-MS analysis and identification of CAP degradation products catalyzed by biocathode |
生物阳极反转生物阴极前后电极生物膜的活性变化通过循环伏安法(CV)和交流阻抗(EIS)分析,结果如图 7所示,生物阳极和反转后的生物阴极起峰电位高于非生物阴极对照,而且峰电流也大于对照,表明二者嗜电极生物膜均具有较强的微生物催化能力. 而3种模式下极化内阻依次为18.87 Ω<31.29 Ω<42.86 Ω,可见生物阳极的极化内阻最低,其次为生物阴极,均明显低于非生物阴极对照42.86 Ω. 电极微生物可以加速阴极电子传递过程具有催化还原降解氯霉素的能力[14],功能电极微生物越多,催化活性越强,催化电流越大,极化内阻值越小. 由此可见生物阳极可能具有最多的嗜电极功能微生物,而反转生物阴极后的电极生物膜由于阴极电位的改变,生物膜的数量和结构可能发生一定的改变,但与非生物阴极相比依然具有较强的生物催化能力.
 | 图 7 生物阳极反转生物阴极前后循环伏安及交流阻抗分析Fig. 7 CV and EIS analysis before and after inversion of bionade to biocathode |
(1)利用无隔膜微生物电催化反应器,采用氯霉素浓度梯度增加的方式成功驯化了能够降解氯霉素的生物阳极,当氯霉素浓度高达80 mg ·L-1时仍然具有较高的降解速率常数(k=0.987 8).
(2)驯化后的生物阳极可以在有隔膜双极室BES反应器中实现生物阳极和非生物阴极同步降解氯霉素,在两个隔室内的还原降解速率常数k依次为0.098 5和0.513 5,该生物阳极不仅具有耐受氯霉素毒性,而且对氯霉素具有一定的降解能力.
(3)利用恒电位仪将生物阳极反转后(此时为生物阴极)恒定在-0.40 V,表现出较好的生物阴极催化还原降解氯霉素的能力,其还原降解速率常数k为0.263 4,远大于非生物阴极对照的速率常数k(0.160 9). 长时间生物阴极的强还原环境使CAP实现了硝基还原、 羰基还原和完全脱Cl.
| [1] | 吴晓丰, 杨鹭花. 氯霉素残留的危害及其检测方法[J]. 动物医学进展, 2004, 25 (3): 41-43. |
| [2] | Martelli A, Mattioli F, Pastorino G, et al. Genotoxicity testing of chloramphenicol in rodent and human-cells[J]. Mutation Research, 1991, 260 (1): 65-72. |
| [3] | Lin A Y C, Yu T H, Lin C F. Pharmaceutical contamination in residential, industrial and agricultural waste streams: Risk to aqueous environments in Taiwan[J]. Chemosphere, 2008, 74 (1): 131-141. |
| [4] | Kasprzyk-Hordern B, Dinsdale R M, Guwy A J. The occurrence of pharmaceuticals, personal care products, endocrine disruptors and illicit drugs in surface water in South Wales, UK[J]. Water Research, 2008, 42 (13): 3498-3518. |
| [5] | Peng X Z, Wang Z D, Kuang W X, et al. A preliminary study on the occurrence and behavior of sulfonamides, ofloxacin and chloramphenicol antimicrobials in wastewaters of two sewage treatment plants in Guangzhou, China[J]. Science of the Total Environment, 2006, 371 (1-3): 314-322. |
| [6] | Xu W H, Zhang G, Li X D, et al. Occurrence and elimination of antibiotics at four sewage treatment plants in the Pearl River Delta (PRD), South China[J]. Water Research, 2007, 41 (19): 4526-4534. |
| [7] | Leung H W, Minh T B, Murphy M B, et al. Distribution, fate and risk assessment of antibiotics in sewage treatment plants in Hong Kong, South China[J]. Environment International, 2012, 42 : 1-9. |
| [8] | Li J, Shao B, Shen J Z, et al. Occurrence of chloramphenicol-resistance genes as environmental pollutants from swine feedlots[J]. Environmental Science & Technology, 2013, 47 (6): 2892-2897. |
| [9] | 刘虹, 张国平, 刘丛强, 等. 贵阳城市污水及南明河中氯霉素和四环素类抗生素的特征[J]. 环境科学, 2009, 30 (3): 687-692. |
| [10] | Xi C W, Zhang Y L, Marrs C F, et al. Prevalence of antibiotic resistance in drinking water treatment and distribution systems[J]. Applied and Environmental Microbiology, 2009, 75 (17): 5714-5718. |
| [11] | Parsley L C, Consuegra E J, Kakirde K S, et al. Identification of diverse antimicrobial resistance determinants carried on bacterial, plasmid, or viral metagenomes from an activated sludge microbial assemblage[J]. Applied and Environmental Microbiology, 2010, 76 (11): 3753-3757. |
| [12] | 徐扣珍, 陆文雄, 宋平, 等. 焚烧法处理氯霉素生产废水[J]. 环境科学, 1998, 19 (4): 69-71. |
| [13] | Mu Y, Rozendal R A, Rabaey K, et al. Nitrobenzene removal in bioelectrochemical systems[J]. Environmental Science & Technology, 2009, 43 (22): 8690-8695. |
| [14] | Liang B, Cheng H Y, Kong D Y, et al. Accelerated reduction of chlorinated nitroaromatic antibiotic chloramphenicol by biocathode[J]. Environmental Science & Technology, 2013, 47 (10): 5353-5361. |
| [15] | Wang A J, Cheng H Y, Liang B, et al. Efficient reduction of nitrobenzene to aniline with a biocatalyzed cathode[J]. Environmental Science & Technology, 2011, 45 (23): 10186-10193. |
| [16] | Liang B, Cheng H Y, van Nostrand J D, et al. Microbial community structure and function of Nitrobenzene reduction biocathode in response to carbon source switchover[J]. Water Research, 2014, 54 : 137-148. |
| [17] | 孙飞, 王爱杰, 严群, 等. 生物电化学系统还原降解氯霉素[J]. 生物工程学报, 2013, 29 (2): 161-168. |
| [18] | Cui D, Guo Y Q, Cheng H Y, et al. Azo dye removal in a membrane-free up-flow biocatalyzed electrolysis reactor coupled with an aerobic bio-contact oxidation reactor[J]. Journal of Hazardous Materials, 2012, 239-240 : 257-264. |
| [19] | Wang Y Z, Wang A J, Zhou A J, et al. Electrode as sole electrons donor for enhancing decolorization of azo dye by an isolated Pseudomonas sp. WYZ-2[J]. Bioresource Technology, 2014, 152 : 530-533. |
| [20] | Strycharz S M, Gannon S M, Boles A R, et al. Reductive dechlorination of 2-chlorophenol by Anaeromyxobacter dehalogenans with an electrode serving as the electron donor[J]. Environmental Microbiology Reports, 2010, 2 (2): 289-294. |
| [21] | Wen Q, Kong F Y, Zheng H T, et al. Simultaneous processes of electricity generation and ceftriaxone sodium degradation in an air-cathode single chamber microbial fuel cell[J]. Journal of Power Sources, 2011, 196 (5): 2567-2572. |
| [22] | Harnisch F, Gimkiewicz C, Bogunovic B, et al. On the removal of sulfonamides using microbial bioelectrochemical systems[J]. Electrochemistry Communications, 2013, 26 : 77-80. |
| [23] | Rozendal R A, Jeremiasse A W, Hamelers H V, et al. Hydrogen production with a microbial biocathode[J]. Environmental Science & Technology, 2008, 42 (2): 629-634. |
| [24] | Cheng K Y, Ho G, Cord-Ruwisch R. Novel methanogenic rotatable bioelectrochemical system operated with polarity inversion[J]. Environmental Science & Technology, 2011, 45 (2): 796-802. |
| [25] | Cheng K Y, Ginige M P, Kaksonen A H. Ano-cathodophilic biofilm catalyzes both anodic carbon oxidation and cathodic denitrification[J]. Environmental Science & Technology, 2012, 46 (18): 10372-10378. |