 2015, Vol. 36
2015, Vol. 36



反硝化过程是氮素由活性氮向惰性氮转化的唯一途径,是实现完整氮循环和全球氮平衡的关键环节[1]. 水体反硝化因此成为减缓水体富营养化的重要途径[2]. 水体反硝化产生的环境效应(产生N2和N2 O)主要表现在两方面:一是反硝化过程可永久性去除氮; 二是反硝化过程产生的N2 O是“京都议定书”规定的主要削减的温室气体之一[3]. 全球氮负荷不断增加已极大促进了水生系统的反硝化进程及N2 O释放通量. 河口区是氮素环境生物地球化学反应的敏感区,存在强烈的反硝化过程. 河口区反硝化作用对水体富营养化控制及全球气候变化具有重要意义. 基于全球尺度的研究表明,河口区约50%的氮素可通过反硝化作用去除[4]; 同时,至2050年河流和河口区N2 O排放量将占全球排放总量的25%[5]. 因此,河口区反硝化速率与N2 O源汇机制研究得到了国内外学者的格外关注. 目前,国际上多以沉积物水界面或水气界面氮释放通量方式揭示河口区反硝化速率大小[6],而直接报道反硝化氮损失比例的研究并不多见. 另外,当前在水体反硝化发生机制方面的认识也存在误区和研究的不足. 传统观点认为水体低氧或厌氧为反硝化发生的必要条件[7]. 而随着水生生态系统好氧反硝化细菌的发现,人们逐渐认识到高氧条件下反硝化过程可能依然存在,且好氧与厌氧反硝化过程的主要产物有所差异[8]. 因此,河口区反硝化过程的受控机制仍有待于进一步研究讨论.
近些年我国主要河口区均呈现氮污染加剧的态势,但国内对河口区反硝化作用的富营养化调控及其温室效应研究还较为缺乏. 本研究以我国典型北方河口区——大辽河口为例,对反硝化过程的环境效应及影响因素进行了探讨,以期为揭示反硝化对河口区富营养化的缓冲作用研究提供借鉴,同时为准确制定我国温室气体排放清单提供参数支持. 1 材料与方法 1.1 研究区概况及采样站位设置
大辽河由浑河和太子河汇合而成,在辽宁省营口市流入辽东湾. 大辽河口位于辽东湾北岸,属典型北方河口区. 大辽河河口区水文状况较为复杂,除受大辽河水系影响外,主要受潮汐控制. 大辽河流域涉及抚顺、 沈阳、 鞍山、 本溪、 辽阳、 盘锦、 营口等7个城市,是辽宁省经济较为发达的地区. 随着流域社会经济的发展,大辽河已成为辽东湾氮、 磷等营养盐的主要输入河流. 研究于2013年夏季(8月)对大辽河干流及其河口区开展了现场采样调查. 为体现淡水(S<0.5‰)和咸水(S>0.5‰)端反硝化作用的差异,调查共设40个采站位; 其中大辽河干流设置8个采站位,从上游至下游依次为R1~R8. 河口区设置32个采站位,依次为E1~E32. 采样站位如图 1所示.
 | 图 1 大辽河及其河口区反硝化调查采样站位示意
Fig. 1 Map of Daliao River estuary and sampling sites
|
采用卡盖式采水器采集表层水样(距水面约0.4 m). 测定溶解N2和N2 O的水样用硅胶管分别引流至10 mL具塞比色管和60 mL血清瓶. 引流时将硅胶管深入比色管和血清瓶底部,通过控制引流速度使水样流出时不产生漩涡和气泡. 当水样溢出比色管和血清瓶二分之一体积时,停止引流,并分别向比色管和血清瓶内水样滴加饱和氯化锌溶液灭活,后旋紧瓶盖密封; 密封后保证水样中没有气泡产生,否则重新采集样品[9]. 同时分装水样于500 mL聚乙烯瓶测定氮、 磷等(TN、 NO-3、 NH+4、 TP、 DTP、 PO3-4)营养盐浓度. 干流氮、 磷营养盐指标的样品采集、 保存方法参照地表水国标方法进行. 河口区氮、 磷营养盐指标的样品采集及保存方法参照《海洋监测规范》进行. 其它水质指标,如pH、 DO、 盐度、 水温、 电导率等利用YSI6600V2现场直接测定. 现场风速用手持风速仪(FC-856)测定,测定高度为距海面2 m. 1.3 室内分析
溶存N2用膜进样质谱仪(MIMS,Bay Instruments,Easton,MD,USA)进行测定,分析测试方法参考文献[10]. 测定溶存N2 O时,首先利用“顶空平衡法”对水样进行处理[11],后用HP5890Ⅱ气相色谱测定顶空气体N2 O浓度(单位×10-9),最后根据顶空N2 O浓度换算水体溶存N2 O浓度(单位 mol ·L-1)[12]. TN、 NO-3、 NH+4、 TP、 DTP、 PO3-4浓度利用营养盐分析仪(QAATRO)测定. 1.4 水-气界面N2及N2 O交换通量计算
水-气界面N2及N2 O交换通量[F,mmol ·(m2 ·d)-1]计算公式为:
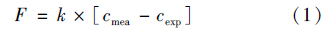
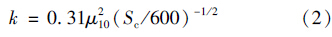
Sc为气体的黏滞系数,N2及N2 O的Sc计算公式见式(3)和式(4):
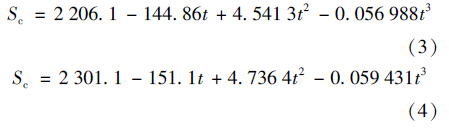
受条件限制,研究实测了距水面2 m高处风速,因此本文采用其它研究中的经验公式对10 m高处的瞬时风速进行了估算,估算公式如下[14]:

研究区域水体盐度呈现出明显的空间分布特征. 大辽河干流站位(R1~R8)盐度变化范围介于0.01‰~0.23‰,为淡水区; 而河口区(E1~E32)盐度变化范围介于1.61‰~23.19‰,属咸水区,且伴随站位向口门外部延伸盐度递增[图 2(a)]. 干流水体DO浓度介于4.38~5.56 mg ·L-1(均值4.66 mg ·L-1),而河口区DO浓度则显著高于干流(P<0.001),其变化范围为3.86~7.67 mg ·L-1(均值5.90 mg ·L-1). 干流氮、 磷营养盐浓度显著高于河口区. 干流TN、 NO-3、 NH+4的平均浓度分别为3.55、 2.29和0.59 mg ·L-1,河口区的浓度均值分别为2.92、 1.35和0.38 mg ·L-1[图 2(b)]; 大辽河干流及河口区氮的主要赋存形态为溶解态,所占比例分别为81%和59%. 干流TP、 DTP、 PO3-4的平均浓度分别为0.24、 0.07和0.06 mg ·L-1,河口区浓度均值为0.16、 0.07和0.06 mg ·L-1 [图 2(c)]; 溶解态磷仍为磷的主要赋存形态,且在干流和河口区的存在比例分别为54%和81%.
 | 图 2 大辽河干流及河口区盐度及氮、 磷浓度
Fig. 2 Salinity,concentrations of N and P in the Daliao River and estuary
|
溶解N2的实测浓度介于460.58~481.45 μmol ·L-1,现场温、 盐条件下N2的理论平衡浓度介于409.77~479.01 μmol ·L-1; ΔN2的负值均出现在河道干流区及部分河口区(E1,S=1.61‰; E2,S=3.91‰)(-11.01~-1.65 μmol ·L-1). 而河口区E3~E32站位(S>7‰)ΔN2均为正增量(2.20~71.37 μmol ·L-1)[图 3(a)]. N2 O实测溶存浓度介于14.69~114.85nmol ·L-1,理论平衡浓度的变化幅度较小,整体上介于7.41~7.62 nmol ·L-1; 由此计算得到ΔN2 O介于7.21~107.33 nmol ·L-1[图 3(b)],N2 O饱和度介于196%~1 528%. 与溶解N2不同的是,干流区ΔN2 O显著高于河口区. 如不考虑硝化及其他N2 O产生过程的影响,河口区N2 O占反硝化总产物(N2 O+N2)的比例为0.007%.
 | 图 3 大辽河及河口区溶存N2 O、 N2净增量
Fig. 3 Net increase of dissolved N2 O and N2 in the Daliao River and estuary
|
距水面2 m高处实测风速及10 m高处估算风速的变化范围分别为0.5~8.7 m ·s-1和0.65~11.34 m ·s-1. 由此估算得到水气界面N2交换通量介于-169.1~1 342.1 mmol ·(m2 ·h)-1,干流河道N2均为负通量. 水气界面N2 O交换通量介于0.018~13.7 mmol ·(m2 ·h)-1,表明研究区域为大气N2 O的释放源. 与N2 O溶存浓度对应,大辽河干流N2 O通量显著高于河口区. 河口区N2 O通量占总释放通量(N2 O+N2)的比例为0.01%~0.77%,均值为0.04%. 3 讨论 3.1 河口区营养盐浓度变化及其原因
大辽河流干流营养盐浓度主要受人类活动影响,如农业地表径流和点源排放等. 而河口区除近岸水体受人类活动影响较大外,离岸区域营养盐的时空异质性主要受水体交换、 有机物降解、 藻类生物吸收利用、 大气沉降等自然过程的影响. 本研究中,大辽河干流氮、 磷浓度显著高于河口区. 水体氮、 磷与盐度数据的相关性分析显示,干流区TN、 NO-3、 NH+4、 TP、 DTP、 PO3-4浓度与盐度完全不相关(大辽河为感潮河流,干流水体仍监测到一定的盐度,但盐度均低于0.5‰,属淡水),见图 4; 而河口区氮、 磷浓度均与盐度表现出显著的线性负相关,且中低盐度区(近岸水体区域)氮、 磷浓度与盐度表现出一定的离散性. 河口区NH+4浓度与盐度的相关性相对较低,表明NH+4浓度除受海水稀释外还受到其它理化过程的影响. 河口区是营养盐生物地球化学反应的敏感区. 尤其是河口区悬浮物的吸附解吸作用对水体溶解性磷具有缓冲作用; 另外,氮、 磷在由近岸向远岸输送过程中会被多种生物化学过程所消耗; 如微生物和藻类吸收无机氮合成为有机氮、 微生物和藻类等死亡后的生物体中的有机氮也会被分解者分解转化为氨氮,氨氮又可被硝化菌氧化为硝氮,并被继续反硝化转化为氮气等. 这些过程均可以导致河口区营养盐浓度的降低. 因此,河口区氮、 磷浓度的递减及其与盐度的显著负相关,并不能完全解释为冲淡水扩散和海水稀释的影响. 本研究中E2、 E3观测站位TN、 TP浓度出现一个相对较高的跃点;而对应两个站位水体总颗粒物(TSS)的浓度分别为205 mg ·L-1和180 mg ·L-1(所有样品TSS变化范围7~205 mg ·L-1),分别为所有点位中的最高和次高值; 因此,由于两个点位处于咸淡水交界处,受河流冲击的影响,水体泥沙含量较高,导致TN、 TP浓度相对较高. 整个河口区溶解态氮、 磷的浓度变化幅度很小,这可能是因为氮、 磷的主要赋存形态为溶解态,而河口区TSS含量相对较低,颗粒物对溶解态氮、 磷的吸附量较小且吸附-解吸过程很容易达到平衡所致.
 | 图 4 河口区氮、 磷浓度与盐度相关性分析
Fig. 4 Correlations of N,P concentrations and salinity
n=32,显著性检验水平为0.05 |
本研究对ΔN2与氮浓度、 DO、 水温、 盐度等环境因子进行了相关分析,以讨论反硝化过程的主要影响因素. 淡水区ΔN2为负值,因此未对此部分数据进行相关分析. 但这并不表明河道干流不存在反硝化过程. 导致这种现象的原因有可能是水体中发生了强烈的固氮作用; 在水体反硝化作用较弱的情况下(固氮速率高于反硝化速率),水体中大量N2被固氮作用消耗,导致水体ΔN2为负值. 这也提示人们,在今后研究河流及河口区反硝化速率时,必须要考虑水体固氮作用带来的误差和影响. 咸水区ΔN2与DO不相关,而与水温和盐度显著正相关,与NO-3显著负相关(表 1). 这种结论尤其值得关注. 传统意义上,反硝化为严格的厌氧过程,DO水平会显著影响反硝化进程[15]. 造成本研究结论的原因之一可能是研究采集的为表层水样(0.4 m),样品采集过程中会对水体产生扰动,水体DO测定浓度存在一定误差(数值偏高),从而导致ΔN2与DO不相关. 另外一种可能是,水生生态系统中存在好氧反硝化细菌,并且某些好氧反硝化细菌对DO水平的响应并不敏感; 在低DO浓度条件下,好氧反硝化细菌参与的反硝化速率较高; 但DO浓度较高时,反硝化过程依然存在,甚至在一定DO水平区间反硝化速率不会发生明显改变[16, 17]. 一般来说,随着水体盐度升高反硝化细菌活性受抑,反硝化速率相应降低[18]. 如Kemp等[19]的研究表明,盐度上升时河口沉积物中硝化活动逐渐降低,与硝化过程耦合的反硝化活动也随之下降. 但也有研究表明,盐度显著影响着硝化而不是反硝化过程. 当盐度在0~15‰之间时,盐度越高硝化速率也越高. 因此,从目前的研究结果看盐度对反硝化速率的影响机制仍不十分明确,一些结论还存在争议[20]. 作者认为,河口区反硝化过程发生的主要源区为沉积物,虽然水体中也存在反硝化过程,但反硝化速率较低. 水体中累积的溶存N2主要受沉积物贡献影响. 因此,本研究河口区水体ΔN2与盐度正相关,可能是由于离口门位置越远(盐度增加),沉积物厚度及有机质含量增加,沉积物中溶解氧的渗透层变浅,反硝化速率增加导致水体N2累积浓度增加所致. 另外,河口区ΔN2与NO-3显著负相关,表明水体反硝化过程为硝化-反硝化耦合作用机制. 其原因有二:一是水体并非厌氧环境,有硝化过程发生的必要条件; 二是河口区水体NO-3浓度受外源输入影响较小,反硝化过程参与的NO-3可能主要来源于硝化过程. 因此NO-3浓度逐渐降低,对应的水体ΔN2增加[图 3(a)],二者显著负相关.
| 表 1 河口区ΔN2净增量与环境因子相关性分析 Table 1 Correlation analysis of ΔN2 net increase and environmental factors |
反硝化可分为化学和生物反硝化两种过程,而化学反硝化过程只存在于深层土壤或者海底沉积物等系统[21]. 因此,本研究所说的反硝化过程一般指生物反硝化过程,是反硝化细菌在一定条件下将活性氮(NO-3)转换为惰性氮(N2 O、 N2)的过程. 同时,反硝化作用是氮的形态转化对环境变化的一种反馈机制,受控于营养盐的组成和水化学条件的变动. 因此反硝化细菌对环境条件的响应控制着反硝化过程的发生及其速率,单纯从环境化学角度已经很难来定性判断反硝化过程的主要受控机制. 因此,从环境微生物学角度解析反硝化的发生机制和生境条件,明确反硝化作用的时空变化及影响因素应成为后续研究的重点关注领域. 3.3 N2 O释放系数
全球陆地系统活性氮负荷不断增加,甚至已趋于饱和. 过量的氮素随地表径流、 下渗等过程输入河流及河口区,并产生大量的N2 O释放到大气. 如Beaulieu等[22]的研究表明,全球范围内输入河流中的DIN约0.75%可转化成N2 O释放到大气. 有估算表明,至2050年大气中约25%的N2 O将来源于河流和河口区的排放[5]. 因此,河流及河口区N2 O的产生及释放无论是作为氮素生物地球化学循环的重要环节还是对全球温室效应的贡献机制均受到了格外关注. 反硝化过程同样可产生N2 O. 本研究中无论淡水区还是咸水区ΔN2 O与盐度、 NO-3、 DO、 水温等环境因子均未表现出显著的相关性(表 2). 这在一定程度上反映了水体中N2 O的产生机制较为复杂,来源并不单一.
| 表 2 ΔN2 O净增量与环境因子相关性分析 Table 2 Correlation analysis of ΔN2 O net increase and environmental factors |
IPCC提供了地下水和地表沟渠(EF5-g)、 河流(EF5-r)、 河口区(EF5-e)等水生系统N2 O排放系数的建议值[23]. 对于河流系统EF5-r最初的建议值(以N2 O/N计,下同)为0.007 5 kg ·kg-1,EF5-e为0.002 5 kg ·kg-1. 很多研究证实,IPCC的排放系数可能高估了全球河流系统N2 O 的排放量,因此2006年EF5-r修改为0.002 5,而EF5-e未做修改. 本研究中EF5-r和EF5-g的实测值([N2 O]/[DIN])分别为0.000 7(0.000 5~0.001 1)和0.000 4(0.000 2~0.001 8),即河流和河口区分别有0.07%和0.04%的DIN转化为N2 O释放到大气; 且二者均低于IPCC的建议值. 因此,不同地理区域及环境条件下,EF5-r及EF5-g的普遍适用性仍需进一步研究讨论. 3.4 河口区反硝化过程脱氮比例
反硝化对河口区富营养化的调控及缓解作用,国际上已展开了一些研究. 这些研究讨论主要针对反硝化过程对水体氮负荷的削减比例来开展. 河口区反硝化的除氮比例又主要借助时空尺度上反硝化速率或水气界面气态氮释放通量等参数进行估算,或者采用物质平衡法、 氮气通量法和同位素示踪技术等手段实现[24]. 作者认为,反硝化过程的主要产物N2和N2 O为惰性气体,而N2 O的产生比例相对很低(<1%),因此水体中N2净增量与氮浓度比例可直接反映反硝化的脱氮系数. 借助这种思路,作者计算得到大辽河河口约26%的TN、 37%的DIN和43%的NO-3会通过反硝化过程得以去除(图 5).
 | 图 5 大辽河河口反硝化氮损失比例
Fig. 5 Percentage of nitrogen loss by denitrification in the Daliao estuary
|
由于河口区为开放性水体,一部分N2会释放到大气,因此考虑到水气界面气态氮的释放,反硝化过程对大辽河河口区的脱氮比例会更高. 国内有关河口区反硝化氮损失比例的研究并不多见. Wu等[25]采用水-气界面N2及N2 O释放通量和水域面积等参数估算了九龙江河口区反硝化氮损失比例; 其结果表明,约24%的DIN通过反硝化过程得以去除. 而作者对夏季渤海湾的调查结果显示,水体反硝化对氮(TN)的损失比例只有1.6%(未发表数据). 这种区域性差异在国际上已有的研究中也有所体现. 如研究表明,输入到波罗的海的总氮约有74%可通过反硝化过程去除,而Norsminde湾反硝化过程对氮的损失比例却只有2%(图 6). 虽然这些水体反硝化过程对氮素的去除比例差异有可能是测定或估算方法带来的误差,但从本研究结果看反硝化对富营养化水体的调控作用仍不可忽视. 在水体富营养化程度不断加剧及全球变化背景下,反硝化过程对水体富营养化的缓冲和控制作用有待于更深入的追踪研究.
 | 数据主要来源于文献[20,25~29]; BS:Baltic Sea、 BH:Boston Harbor、 CB:Chesapeake Bay、 DB:Delaware Bay、 NB:Narragansett Bay、 OB:Ochlockonee Bay、 NF:Norsminde Fjord、 PR:Potomac River、 SE:Scheldt Estuary、 DE:大辽河河口区、 BB:渤海湾、 JE:九龙江河口区; 九龙江河口区数据为DIN损失比例 图 6 不同区域水体反硝化氮损失比例 Fig. 6 Percentage of nitrogen loss by denitrification in various aquatic systems |
(1)大辽河干流水体氮、 磷浓度高于河口区. 河口区氮、 磷浓度与盐度表现出较好的保守性; 河口区相对于干流氮、 磷浓度的降低主要受海水稀释和其它生物地球化学过程的共同影响.
(2)大辽河河口区存在明显的反硝化过程,河口区约26%的TN、 37%的DIN和43%的NO-3通过反硝化过程得以去除. 反硝化过程中N2 O的产生比例平均为0.007%,而N2 O释放通量占总通量的比例为0.04%. 本研究EF5-r和EF5-g实测值([N2 O]/[DIN])分别为0.000 7和0.000 4,均低于IPCC的建议值.
(3)大辽河河口区ΔN2与DO不相关,而与水温和盐度显著正相关,与NO-3显著负相关; 表明反硝化过程不完全受控于DO水平,且河口区的主要反硝化机制为耦合硝化-反硝化过程. 与传统研究相比,本研究中反硝化过程在高DO水平下仍然存在,并且盐度越高反硝化过程越强烈,因此建议加强河口区反硝化细菌种类及从环境微生物角度解释影响反硝化作用的研究.
致谢: 感谢中国科学院南京土壤研究所颜晓元研究员和赵永强博士对溶存N2测定所提供的帮助. 感谢李拴虎、 王雪纯、 林暾、 蔡林颖等在样品采集和分析方面提供的帮助.
| [1] | Murray R E, Knowles S R. Production of NO and N2O in the presence and absence of C2H2 by soil slurries and batch cultures of denitrifying bacteria[J]. Soil Biology and Biochemistry, 2003, 35 (8): 1115-1122. |
| [2] | Seitzinger S P, Harrison J A, Bohlke J K, et al. Denitrification across landscapes and waterscapes: a synthesis[J]. Ecological Applications, 2006, 16 (6): 2064-2090. |
| [3] | Ravishankara A R, Daniel J S, Portmann R W. Nitrous oxide (N2O): The dominant ozone-depleting substance emitted in the 21st Century[J]. Science, 2009, 326 (5949): 123-125. |
| [4] | Howarth R W, Billen G, Swaney D, et al. Regional nitrogen budgets and riverine N and P fluxes for the drainages to the North Atlantic Ocean-Natural and human influences[J]. Biogeochemistry, 1996, 35 (1): 75-139. |
| [5] | Seitzinger S P, Kroeze C. Global distribution of nitrous oxide production and N inputs in freshwater and coastal marine ecosystems[J]. Global Biogeochemical Cycles, 1998, 12 (1): 93-113. |
| [6] | 王东启, 陈振楼, 许世远, 等. 长江口潮滩沉积物反硝化作用及其时空变化特征[J]. 中国科学(B辑): 化学, 2007, 37 (6): 604-611. |
| [7] | Patrick W H, Reddy K R. Nitrification-Denitrification reactions in flooded soils and water bottoms: dependence on oxygen supply and ammonium diffusion[J]. Journal of Environmental Quality, 1976, 5 (4): 469-471. |
| [8] | 李平, 张山, 刘德立. 细菌好氧反硝化研究进展[J]. 微生物学杂志, 2004, 25 (1): 60-64. |
| [9] | 陈能汪, 吴杰忠, 段恒轶, 等. N2:Ar 法直接测定水体反硝化产物溶解N2[J]. 环境科学学报, 2010, 30 (12): 2479-2483. |
| [10] | 李晓波, 夏永秋, 郎漫, 等. N2:Ar 法直接测定淹水环境反硝化产物 N2的产生速率[J]. 农业环境科学学报, 2013, 32 (6): 1284-1288. |
| [11] | Johnson K M, Hughes J E, Donaghay P L, et al. Bottle-calibration static head space method for the determination of methane dissolved in seawater[J]. Analytical Chemistry, 1990, 62 (21): 2408-2412. |
| [12] | 杨丽标, 王芳, 晏维金. 巢湖流域河流沉积物N2O释放对水体溶存N2O贡献研究[J]. 农业环境科学学报, 2013, 32 (4): 771-777. |
| [13] | Weiss R F. The solubility of nitrogen, oxygen and argon in water and seawater[J]. Deep-Sea Research and Oceanographic Abstracts, 1970, 17 (4): 721-735. |
| [14] | Israelsen O W, Hansen V E. Irrigation principles and practices[M]. New York: John Wiley & Sons Inc., 1962. |
| [15] | Tiedje J M. Ecology of denitrification and dissimilatory nitrate reduction to ammonium[A]. In: Zehnder A J B (Ed.). Biology of anaerobic microorganisms[M]. New York: John Wiley and Sons, 1988. |
| [16] | Patureau D, Bernet N, Delgenès J P, et al. Effect of dissolved oxygen and carbon nitrogen loads on denitrification by an aerobic consortium[J]. Applied Microbiology and Biotechnology, 2000, 54 (4): 535-542. |
| [17] | 胡朝松, 李春强, 刘志昕, 等. 海洋沉积物中反硝化细菌的分离鉴定及反硝化性能研究[J]. 环境科学研究, 2009, 22 (1): 114-118. |
| [18] | Rysgaard S, Thastum P, Dalsgaard T, et al. Effects of salinity on NH4+ adsorption capacity, nitrification, and denitrification in Danish Estuarine sediments[J]. Estuaries, 1999, 22 (1): 21-30. |
| [19] | Kemp W M, Sampou P, Caffrey J, et al. Ammonium recycling versus denitrification in Chesapeake Bay sediments[J]. Limnology and Oceanography, 1990, 35 (7): 1545-1563. |
| [20] | Cornwell J C, Kemp W M, Kana T M. Denitrification in coastal ecosystems: methods, environmental controls, and ecosystem level controls, a review[J]. Aquatic Ecology, 1999, 33 (1): 41-54. |
| [21] | Christianson C B, Cho C M. Chemical denitrification of nitrite in frozen soils[J]. Soil Science Society of America Journal, 1983, 47 (1): 38-42. |
| [22] | Beaulieu J J, Tank J L, Hamilton S K, et al. Nitrous oxide emission from denitrification in stream and river networks[J]. Proceedings of the National Academy of Sciences of the United States of America, 2011, 108 (1): 214-219. |
| [23] | Intergovernmental Panel on Climate Change (IPCC). IPCC Guidelines for National Greenhouse Gas Inventories, Prepared by the National Greenhouse Gas Inventories Programme[A]. In: Eggleston H A, Buendia L, Miwa K (Eds.). Institute for Global Environ[C]. Strategies, Hayama, Kanagawa, Japan: IPCC/IGES, 2006. |
| [24] | 席北斗, 于会彬, 马文超, 等. 湖岸缓冲带反硝化作用的研究进展[J]. 环境工程学报, 2009, 3 (10): 1729-1734. |
| [25] | Wu J Z, Chen N W, Hong H S, et al. Direct measurement of dissolved N2 and denitrification along a subtropical river-estuary gradient, China[J]. Marine Pollution Bulletin, 2013, 66 (1-2): 125-134. |
| [26] | Nixon S W, Ammerman J W, Atkinson L P, et al. The fate of nitrogen and phosphorus at the land-sea margin of the North Atlantic Ocean[J]. Biogeochemistry, 1996, 35 (1): 141-180. |
| [27] | Nielsen K, Nielsen L P, Rasmussen P. Estuarine nitrogen retention independently estimated by the denitrification rate and mass balance methods: a study of Norsminde Fjord, Denmark[J]. Marine Ecology Progress Series, 1995, 119: 275-283. |
| [28] | Giblin A E, Hopkinson C S, Tucker J. Benthic metabolism and nutrient cycling in Boston Harbor, Massachusetts[J]. Estuaries, 1997, 20 (2): 346-364. |
| [29] | Seitzinger S P and Giblin A E. Estimating denitrification in North Atlantic continental shelf sediments[J]. Biogeochemistry, 1996, 35 (1): 235-260. |