 2015, Vol. 36
2015, Vol. 36


2. 河北科技大学环境科学与工程学院, 河北省污染防治生物技术重点实验室, 石家庄 050018
2. Pollution Prevention Biotechnology Laboratory of Hebei Province, School of Environmental Science and Engineering, Hebei University of Science and Technology, Shijiazhuang 050018, China
臭氧氧化是一种高效水处理技术,几乎无二次污染[1]. 然而,传统臭氧处理技术存在气液传质速率慢、 氧化能力弱和臭氧利用效率偏低等缺点,因此提高臭氧利用效率和氧化能力是臭氧处理技术应用的关键问题[2,3,4]. 微气泡通常是指直径为10-50 μm的微小气泡,与传统气泡相比,微气泡有助于提高气液传质速率和效率[5,6]; 同时微气泡在液相中具有收缩和破裂特性,并在此过程中产生 ·OH,从而提高其氧化能力[7,8,9]. 微气泡技术在废水处理领域中表现出潜在的应用优势,逐渐受到关注.
采用微气泡与臭氧氧化技术相结合能够强化臭氧传质,提高臭氧利用率,并促进 ·OH产生,显著改善臭氧氧化效果. Shin等[10]发现微气泡臭氧传质速率是传统气泡的2.8倍,且液相臭氧浓度更高,有利于污染物的去除. Chu等[11,12]的研究也证实微气泡臭氧传质系数比传统气泡高1.8倍; 且微气泡系统臭氧利用率始终保持在99%以上,逸气臭氧浓度仅为0.26-1.42 mg ·L-1,而传统气泡系统中逸气臭氧浓度高达2-32.2 mg ·L-1.
本研究采用臭氧微气泡处理酸性大红3R废水,考察了臭氧微气泡气液传质特性、 酸性大红3R氧化处理效果以及氧化反应进程,并与臭氧传统气泡处理过程进行比较,以期为臭氧微气泡技术在废水处理中的实际应用提供参考. 1 材料与方法 1.1 实验装置
实验装置如图 1所示. 以纯氧为气源,采用臭氧发生器(济南景天)产生臭氧气体,通过流量计控制气体流量,并采用碘量法对臭氧产量进行测定. 臭氧与反应器中循环液体进入微气泡发生装置(北京本洲纳米科技有限公司)混合,气-液混合物通过射流器进入反应器底部,产生臭氧微气泡. 在气体流量为0.3 L ·min-1时,通过显微观察、 测量和统计分析[11],测定臭氧微气泡的平均直径为51.4 μm. 同时,采用砂滤头产生臭氧传统气泡. 尾气中的臭氧采用碘化钾溶液进行吸收. 反应器密封,有效容积为10 L.
 | 图 1 实验装置示意Fig. 1 Experimental apparatus |
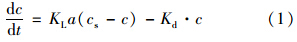
将边界条件t=0、 c=0代入(1)并积分得:
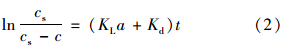
停止曝气后,臭氧在水中分解速率按照式(3)进行计算:

将边界条件t=0、 c=cs代入式(3)并积分得:

本研究采用微气泡和传统气泡两种方式向清水中通入臭氧,控制气体流量为0.3 L ·min-1,此时臭氧投加剂量为48.3 mg ·min-1. 在此条件下,采用靛蓝法测定曝气过程中不同时刻t溶解臭氧浓度c以及饱和臭氧浓度cs. 溶解臭氧浓度达到饱和后停止曝气,并继续测定溶解臭氧浓度c随时间t的变化.
曝气过程中得到ln(cs-c)和t的直线关系,其斜率即为臭氧的总体积传质系数与臭氧分解系数之和(KLa+Kd). 停止曝气后得到ln(cs/c)和t的直线关系,其斜率即为臭氧分解系数Kd[13]. 1.3 臭氧氧化处理酸性大红3R
反应器中配制酸性大红3R废水,浓度约为100 mg ·L-1. 采用臭氧微气泡和传统气泡分别对酸性大红3R废水进行处理,控制气体流量为0.3 L ·min-1,此时臭氧投加剂量为48.3 mg ·min-1. 测定处理过程中的脱色率、 TOC去除率、 溶液pH值和溶液紫外-可见吸收光谱,同时检测液相臭氧浓度和臭氧散逸量随时间变化. 1.4 检测方法
采用紫外-可见分光光度计(上海天美,U-3900)测定酸性大红3R溶液在510 nm处的吸光度变化,并计算脱色率,同时在190-600 nm范围内扫描酸性大红3R溶液的紫外-可见吸收光谱. TOC采用TOC分析仪(日本岛津,TOC-VCPH)测定,pH值采用酸度计(上海雷磁,pHs-3c)进行测定. 液相臭氧浓度采用靛蓝法测定[14],气相臭氧浓度采用碘量法测定[15]. 2 结果与讨论 2.1 臭氧气液传质
臭氧微气泡通入反应器后液相呈现白色乳浊状,表明大量微气泡形成. 停止曝气后,由于微气泡上升速度较慢,液相白色乳浊状可保持3-4 min. 在臭氧投加量48.3 mg ·min-1、 气流量为0.3 L ·min-1的条件下,测定了臭氧微气泡曝气(30min)与传统气泡曝气(60 min)中及停止曝气后,液相臭氧浓度随时间的变化,结果如图 2所示. 可以看到,曝气过程中液相臭氧浓度均迅速上升,微气泡曝气20 min时臭氧浓度达到饱和(5.45 mg ·L-1),传统气泡曝气50 min时臭氧浓度达到饱和(5.66 mg ·L-1). 停止曝气后,由于臭氧自身分解,液相臭氧浓度逐渐降低. 根据此过程中液相臭氧浓度随时间的变化,计算微气泡曝气臭氧传质系数与分解系数分别为0.243 min-1和0.068 min-1,传统气泡曝气臭氧传质系数与分解系数分别为0.067 min-1和0.011 min-1. 微气泡曝气臭氧传质系数与分解系数分别为传统气泡曝气的3.6和6.2倍. 可见,微气泡曝气可以强化臭氧的气液传质过程. 同时,微气泡曝气中臭氧分解速度更快,此过程有利于 ·OH产生进而提高臭氧处理氧化能力.
 | 图 2 臭氧微气泡和传统气泡曝气中液相臭氧浓度 Fig. 2 Dissolved ozone concentration in microbubble aeration and conventional bubble aeration |

以ln(c0/c)为纵坐标,以t为横坐标作图,斜率即为一级反应动力学常数[16],经计算其值分别为0.146 min-1和0.056 min-1. 由于臭氧能够直接氧化破坏酸性大红3R分子中的发色基团偶氮键(—N N—),因而臭氧微气泡与传统气泡氧化处理过程均可以实现100%的脱色率. 微气泡对臭氧气液传质的强化作用是其脱色速率较快的主要原因.
 | 图 3 臭氧微气泡和传统气泡氧化处理酸性大红3R中脱色率 Fig. 3 Color removal efficiency in ozonation treatment of acid red 3R wastewater with microbubble aeration and conventional bubble aeration |
相同条件下臭氧微气泡与传统气泡氧化处理酸性大红3R中TOC去除率变化如图 4所示. 可以看到,臭氧微气泡氧化处理120 min时的TOC去除率为78.0%,远高于臭氧传统气泡的36.2%. 可见,臭氧微气泡显著改善臭氧氧化处理效果. 其原因是臭氧直接氧化对TOC的去除能力有限,TOC去除主要依赖于臭氧分解产生 ·OH; 微气泡曝气中臭氧分解速率更快,能够产生更多 ·OH,从而提高氧化能力.
 | 图 4 臭氧微气泡和传统气泡氧化处理 酸性大红3R中TOC去除率 Fig. 4 TOC removal efficiency in ozonation treatment of acid red 3R wastewater with microbubble aeration and conventional bubble aeration |
在臭氧微气泡氧化处理酸性大红3R过程中加入Na2CO3(1.5 g ·L-1),其TOC去除率变化如图 5所示. 可以看到,投加Na2CO3后微气泡系统TOC去除率显著下降,处理120 min时的TOC 去除率由78.0%下降至47.4%. 而在传统气泡系统中,Na2CO3对TOC去除影响不大,处理120 min时,未投加和投加Na2CO3的TOC 去除率分别为39.5%和41.9%. 可见,Na2CO3作为 ·OH捕获剂,对臭氧微气泡氧化去除TOC能力具有显著影响,证实臭氧微气泡对TOC的高效去除主要是 ·OH作用. 而臭氧传统气泡系统中以臭氧直接氧化为主,因而Na2CO3的影响不大.
 | 图 5 投加Na2CO3后臭氧微气泡和传统气泡 氧化处理酸性大红3R中TOC去除率 Fig. 5 TOC removal efficiency in ozonation treatment of acid red 3R wastewater with microbubble aeration and conventional bubble aeration after addition of Na2CO3 |
臭氧微气泡与传统气泡氧化处理酸性大红3R中液相臭氧浓度和臭氧散逸量变化情况如图 6所示. 可以看到,臭氧传统气泡处理过程中,0-70 min内为传质控制过程,臭氧溶解后即迅速参与反应,液相臭氧浓度从0.08 mg ·L-1缓慢升高至0.45 mg ·L-1. 70 min后随着难以臭氧直接氧化的中间产物的产生和累积,液相臭氧不再参与反应而逐渐累积,液相臭氧浓度迅速升高,120 min时提高至4.12 mg ·L-1,处理过程由传质控制转变为反应控制. 可见,臭氧传统气泡处理以臭氧直接氧化为主.
 | 图 6 臭氧微气泡和传统气泡氧化处理酸性大红 3R中液相臭氧浓度和臭氧逸散量 Fig. 6 Output ozone amount and dissolved ozone concentration in ozonation treatment of acid red 3R wastewater with microbubble aeration and conventional bubble aeration |
臭氧微气泡处理过程中,液相臭氧浓度始终低于0.4 mg ·L-1,表明该过程为传质控制过程,臭氧溶解后即迅速直接参与反应或分解产生 ·OH参与反应. 考虑到臭氧微气泡处理中,脱色速率和TOC去除率均显著高于臭氧传统气泡处理,因此臭氧分解产生 ·OH反应是臭氧微气泡处理的关键反应过程.
同时,臭氧微气泡与传统气泡氧化处理酸性大红3R过程中,臭氧逸散量随时间的变化如图 6所示. 可以看到,微气泡系统臭氧散逸量从0缓慢增加至3.0 mg ·min-1,传统气泡系统臭氧散逸量从4.0 mg ·min-1逐渐增加至22.8 mg ·min-1. 根据臭氧投加量、 液相臭氧浓度和臭氧散逸量计算处理过程中的臭氧消耗速率和臭氧利用率[11],微气泡系统臭氧消耗速率从48.2 mg ·min-1降低至44.9 mg ·min-1,相应地臭氧利用率从99.7%降低至92.9%,平均臭氧利用率为97.8%; 传统气泡系统臭氧消耗速率从44.4 mg ·min-1降低至24.6 mg ·min-1,相应臭氧利用率从92.0%降低至50.9%,平均臭氧利用率为69.3%. 可见,微气泡系统中臭氧的传质速率和反应速率很快,因而臭氧散逸量极低,臭氧利用率显著高于传统气泡系统. 2.4 臭氧氧化酸性大红3R 反应进程
臭氧微气泡与传统气泡氧化处理酸性大红3R过程中紫外-可见吸收光谱变化如图 7所示. 酸性大红3R存在3个明显紫外-可见吸收峰,其中370-590 nm处为偶氮键(—N N—)与芳香环共轭结构吸收峰,340 nm处为偶氮键吸收峰,200-250 nm处为芳香环吸收峰. 反应40 min后,两种处理方式下340 nm及370-590 nm处吸收峰均消失,表明酸性大红3R的发色基团被破坏; 同时,200-250 nm处的吸收强度显著下降,表明芳香环结构也存在一定程度的降解. 随后,微气泡系统中200-250 nm处的吸收强度快速下降,反应120 min后吸收峰几乎消失. 而传统气泡系统中200-250 nm处的吸收强度也持续下降但相对较慢,反应120 min后吸收峰仍然存在. 可见酸性大红3R中的芳香环结构被不断降解,且臭氧微气泡对芳香环的氧化降解速率更快.
 | 图 7 臭氧微气泡与传统气泡氧化处理酸性大红3R中紫外-可见吸收光谱 Fig. 7 UV-visible spectra in ozonation treatment of acid red 3R wastewater with microbubble aeration and conventional bubble aeration |
臭氧微气泡与传统气泡氧化处理酸性大红3R中pH值变化如图 8所示. 臭氧传统气泡处理过程中,溶液pH值由7.84持续下降至3.92,其中60-90 min反应阶段溶液pH值下降较快,从5.79降至4.20. 而臭氧微气泡处理70 min后,溶液pH值由7.18下降至5.89,而后缓慢升高,120 min时提高至6.27. 事实上,臭氧处理过程中pH值下降是由于酸性大红3R降解产生小分子有机酸所致[17,18]. 由于臭氧传统气泡对小分子有机酸降解速率较慢[17],特别是在脱色过程完成后的60-90 min反应阶段,芳香环降解产生小分子有机酸的速率加快,造成小分子有机酸积累速率明显高于降解速率,因此pH值下降显著. 而在臭氧微气泡系统中,溶液pH值下降幅度较小,且反应后期pH值还略有升高,表明臭氧微气泡比传统气泡具有更强的氧化能力和更快的小分子有机酸降解速率,因而大大降低了小分子有机酸的积累. 可见,小分子有机酸降解主要依赖于 ·OH反应,而臭氧微气泡比传统气泡产生 ·OH能力更强,因此可以有效降解小分子有机酸,这一过程与TOC去除是一致的.
 | 图 8 臭氧微气泡和传统气泡氧化处理酸性大红3R中pH值 Fig. 8 pH in ozonation treatment of acid red 3R wastewater with microbubble aeration and conventional bubble aeration |
紫外-可见吸收光谱和pH值变化表明,臭氧微气泡与传统气泡氧化处理酸性大红3R的反应进程可以分为3个环节:①氧化破坏发色基团进行脱色,并生成具有芳香环结构的中间产物; ②氧化降解芳香环结构,直至产生小分子有机酸; ③小分子中间产物(包括小分子有机酸)最终矿化. 通过计算累积TOC去除量与累积臭氧消耗量的比值(R)随时间变化关系[11],进一步比较臭氧微气泡与传统气泡氧化处理酸性大红3R的反应进程,结果如图 9所示.
 | 图 9 臭氧微气泡和传统气泡氧化处理酸性大红3R中TOC去除量与臭氧消耗量比值 Fig. 9 Ratio of removed TOC amount to ozone consumption in ozonation treatment of acid red 3R wastewater with microbubble aeration and conventional bubble aeration |
可以看到,反应初期,微气泡与传统气泡系统的R值较低,表明此时的臭氧消耗主要用于脱色反应. 同时,此时微气泡系统的R值低于传统气泡系统,表明此反应阶段微气泡系统中用于氧化脱色反应的臭氧消耗更多,这与微气泡系统脱色速率更快是一致的.
随着氧化脱色过程的完成,传统气泡系统的R值逐渐升高,表明臭氧消耗从脱色反应逐渐转变为芳香环结构降解产生小分子中间产物(包括小分子有机酸)及其矿化反应. 在此过程中,由于臭氧传统气泡对小分子有机酸的矿化能力有限,是TOC去除的限制步骤,因此随着小分子有机酸的产生和积累,反应40 min后R值有所降低并趋于稳定,此时,臭氧分解产生 ·OH并氧化小分子有机酸去除TOC成为消耗臭氧的主要反应,且反应过程中 ·OH产生速率和TOC去除速率基本保持不变. 传统气泡系统的R值最终保持在0.015-0.016 mg ·mg-1,反映了其对小分子有机酸的矿化能力.
臭氧微气泡处理酸性大红3R的反应进程与传统气泡基本相同,但由于臭氧微气泡对小分子中间产物(特别是小分子有机酸)的氧化矿化能力更强,因此微气泡系统的R值持续上升,并在反应100 min后基本稳定. 同样,此时臭氧产生 ·OH并氧化小分子有机酸去除TOC成为消耗臭氧的主要反应,且反应速率基本保持不变. 微气泡系统的稳定R值为0.025-0.026 mg ·mg-1,同样反映了其对小分子有机酸的矿化能力. 可见,臭氧微气泡对小分子有机酸的氧化矿化能力约为传统气泡的1.6倍. 3 结论
(1)微气泡曝气能够提高臭氧的气液传质速率,相同条件下其臭氧传质系数为传统气泡的3.6倍; 同时微气泡系统的液相臭氧分解系数为传统气泡系统的6.2倍,有利于 ·OH产生.
(2)臭氧微气泡可显著提高臭氧氧化效果. 相同条件下,臭氧微气泡处理酸性大红3R的脱色速率和TOC去除速率均明显高于传统气泡系统,其TOC去除率可达78.0%,为传统气泡的2倍.
(3)臭氧微气泡处理酸性大红3R过程中的臭氧利用率显著高于传统气泡. 微气泡系统臭氧散逸量为0-3.0 mg ·min-1,平均臭氧利用率为97.8%; 传统气泡系统臭氧散逸量为4.0-22.8 mg ·min-1,平均臭氧利用率为69.3%.
(4) 臭氧微气泡通过促进 ·OH产生提高臭氧氧化能力,其对中间产物的降解速率更快,其中对小分子有机酸的氧化矿化能力约为传统气泡的1.6倍.
| [1] | 蔡少卿, 戴启洲, 王佳裕, 等. 非均相催化臭氧处理高浓度制药废水的研究[J]. 环境科学学报, 2011, 31 (7): 1440-1449. |
| [2] | Zhang J, Liu J S, Liu C, et al. Characteristics of activated carbon modified with nitric acid and its performance in catalytic ozonation of acid red 3R[J]. Advanced Material Research, 2013, 726-731: 1687-1690. |
| [3] | 张彭义, 余刚, 孙海涛, 等. 臭氧/活性炭协同降解有机物的初步研究[J]. 中国环境科学, 2000, 20 (2): 159-162. |
| [4] | Keura H, Hamasaki S, Tamaki M, et al. Effects of ozone microbubble treatment on removal of residual pesticides and quality of persimmon leaves[J]. Food Chemistry, 2013, 138 (1): 366-371. |
| [5] | Bredwell M D, Worden R M. Mass-transfer properties of microbubbles 1: experimental studies[J]. Biotechnology Progress, 1998, 14 (1): 31-38. |
| [6] | Liu S, Wang Q H, Ma H Z, et al. Effect of micro-bubbles on coagulation flotation process of dyeing wastewater[J]. Separation and Purification Technology, 2010, 71 (3): 337-346. |
| [7] | Takahashi M, Chiba K, Li P. Free-radical generation from collapsing microbubbles in the absence of a dynamic stimulus[J]. Physical Chemistry B, 2007, 111 (6): 1343-1347. |
| [8] | Li P, Takahashi M, Chib K. Degradation of phenol by the collapse of microbubbles[J]. Chemosphere, 2009, 75 (10): 1371-1375. |
| [9] | Li P, Takahashi M, Chiba K. Enhanced free-radical generation by shrinking microbubbles using a copper catalyst[J]. Chemosphere, 2009, 77 (8): 1157-1160. |
| [10] | Shin W T, Mirmiran A, Yiacoumi S, et al. Ozonation using microbubbles formed by electric fields[J]. Separation and Purification Technology, 1999, 15 (3): 271-282. |
| [11] | Chu L B, Xing X H, Yu A F, et al. Enhanced ozonation of simulated dyestuff wastewater by microbubbles[J]. Chemosphere, 2007, 68 (10): 1854-1860. |
| [12] | Chu L B, Yan S T, Xing X H, et al. Enhanced sludge solubilization by microbubble ozonation[J]. Chemosphere, 2008, 72 (2): 205-212. |
| [13] | Kukuzakia M, Fujimotob K, Kaib S, et al. Ozone mass transfer in an ozone-water contacting process with Shirasu porous glass (SPG) membranes-A comparative study of hydrophilic and hydrophobic membranes[J]. Separation and Purification Technology, 2010, 72 (3): 347-356. |
| [14] | Bader H, Hoigné J. Determination of ozone in water by the indigo method[J]. Water Research, 1981, 15 (4): 449-456. |
| [15] | 宋仁元, 张亚杰, 王维一, 等. 水和废水标准检验法[M]. 北京: 中国建筑工业出版社, 1985. 368-370. |
| [16] | 李炳智, 徐向阳, 朱亮. 氯代硝基苯类废水臭氧化动力学和机理[J]. 化工学报, 2008, 59 (8): 2111-2120. |
| [17] | Faria P C C, Órfão J J M, Pereira M F R. A novel ceria-activated carbon composite for the catalytic ozonation of carboxylic acids[J]. Catalysis Communications, 2008, 9 (11-12): 2121-2126. |
| [18] | Goncalves A, Silvestre-Albero J, Ramos-Fernández E V, et al. Highly dispersed ceria on activated carbon for the catalyzed ozonation of organic pollutants[J]. Applied Catalysis B: Environmental, 2012, 113-114: 308-317. |