 2014, Vol. 35
2014, Vol. 35



生物脱氮技术是污水处理中广为应用的方法,部分城市污水中碳源不足影响了脱氮效率. Kuba等[1]研究表明,当进水C/N比低于3.4时,需投加外碳源来保证生物脱氮效果. 实际处理过程中污水厂通过投加甲醇或乙醇而提高脱氮效率的方式,既消耗了有机资源,又增加了水厂的运行费用. 因此,改进传统工艺从而实现高效脱氮,具有重大的现实意义. 与此同时,作为生物反硝化的中间产物,N2 O的产生也不容忽视[2, 3, 4]. N2 O具有高温室效应,其增温潜势是CO2的250倍[5],是近几年来备受关注的研究热点.
催化铁方法,在铁刨花材料上表面镀铜从而形成双金属体系,在中性以及碱性条件下具有比零价铁更大的还原能力[6]. Chi等[7]研究表明,在低pH条件下,Fe0无机还原NO-3-N产物以氨氮为主,高pH条件下,还原产物主要是微量气态N2 O. Huang等[8]发现,在高NO-3负荷下,随着pH的降低,Fe0还原NO-3-N速率与去除率不断增加. 相比之下,零价铁与生物相互作用脱除NO-3方面的研究较少,王子[9]研究表明催化铁与Paracocus反硝化微生物耦合脱氮是可行的,pH=8时脱氮效率接近于直接使用氢气. 催化铁还可以与水中的溶解氧发生反应,降低周围的氧化还原电位,这对于消除体系内溶解氧,使NO-3成为唯一电子受体具有重要的意义.
本研究尝试催化铁与生物耦合,解决低碳氮比条件下反硝酸电子供体不足的问题,从而实现高效脱氮,并考察反硝化阶段N2 O的产生影响. 1 材料与方法 1.1 水质及污泥驯化
污泥驯化采用人工配水,以NaNO3作为反硝化氮源,CH3COONa作为有机碳源,KH2PO4作为磷源,适当加入碳酸氢钠缓冲液以维持体系pH的稳定. 配水中还投加硫酸镁、 牛肉浸膏,酵母浸出液等物质以及适量的微量元素,见表 1. 其中,微量元素由氯化铁、 氯化钴、 碘化钾、 五水硫酸铜、 钼酸钠、 硫酸锌、 氯化锰、 硼酸、 EDTA组成. 碳氮比控制在6左右. 有机负荷(以BOD5/MLSS计,下同)为0.3~0.4 kg ·(kg ·d)-1,总氮负荷为0.076~0.1 kg ·(kg ·d)-1.
| 表 1 营养元素投加表 /mg ·L-1 Table 1 Concentrations of nutrition elements/mg ·L- 1 |
接种污泥取自污水处理厂二沉池回流污泥. 培养前闷曝24 h,养泥反应器采用10 L有机玻璃反应器,反应运行周期为3 h,进水、 搅拌、 沉淀、 排水时间分别为11、 130、 30、 9 min. 由于反应器完全密闭,其溶解氧含量在0.2 mg ·L-1以下. 控制培养温度为20~25℃,污泥浓度在3000~4000 mg ·L-1,HRT为8.25 h,SRT为15 d. 将污泥培养一个月左右将其驯化为反硝化污泥,进出水COD、 NO-3-N、 TN、 Fe3+基本稳定,总氮去除率达99%. 1.2 试验装置与运行方式
试验采用的SBR反应器如图 1所示,有效容积5.5 L,内径19 cm,高32 cm,试验采用两个SBR反应器进行对比,SBR1传统反硝化反应器,SBR2为催化铁与反硝化耦合反应器,内置铁床,铁投加量为60 g ·L-1,反应器运行周期均为3 h,通过时间控制器实现进水,厌氧搅拌,静置沉淀,出水各个阶段. 每次进排水2 L,充水比为0.364. 进水、 搅拌、 沉淀、 排水时间分别为11、 130、 30、 9 min.
批次试验所用污泥均为之前已驯化完毕污泥,不同水平批次试验开展之前,需将所用污泥在去离子水中清洗3次,然后根据需要(不同碳氮比、 不同pH)投加不同浓度的营养物质进行试验.
 | 图 1 序批式活性污泥反应器装置示意
Fig. 1 Schematic diagram of SBR system
|
采用溶解氧、 pH计、 ORP测定仪进行DO、 pH、 ORP的分析测定,NO-3-N、NO-2-N、NH+4-N、 COD采用国家标准方法测定,TN、 TOC采用岛津TOC-L CPH CN200进行测定.
气态N2 O测定采用Agilent公司7890A气相色谱仪,HP-mole毛细管色谱柱测定N2 O. 色谱条件:进样口温度105℃,炉温180℃; ECD检测器温度300℃,载气为95%Ar+5%CH4,所有气体样品均测定3次,结果取平均值.
亚铁离子与总铁离子采用邻菲啰啉分光光度法,上清液中亚铁离子含量通过3000 r ·min-1离心后测得,酸溶后亚铁离子经过1 ∶1盐酸密闭溶解振荡10 h后测得. 2 结果与分析 2.1 催化铁对反硝化效果的影响
零价铁无机反应为纯化学反应,反应效果受多因素影响[10, 11, 12, 13]. 本试验中,催化铁反应体系pH呈中性,溶液中零价铁、 Fe2+与NO-3的无机反应(即不通过微生物作用而直接进行的纯化学反应,下同)进行得十分缓慢,最终对总氮及硝氮的去除效果不明显,均没有超过10%; 催化铁耦合组对总氮的去除与对照组相比差异不大,均为28%~29%; 但对硝氮的转化非常明显,有98.4%硝酸根得到转化,其中有63.7%转化为亚硝酸根,29.7%转化为氧化亚氮及氮气. 对照组硝酸根转化率明显较低,仅为51.2%(图 2).
 | 图 2 3种工艺硝酸根去除及转化
Fig. 2 Nitrate removal and transformation under three conditions
|
 | 图 3 反应2 h后不同形态氮比例
Fig. 3 Proportions of different nitrogen forms after 2 hours reaction
|
图 3为反硝化2 h结束后各组氮形态分析,无机组仍有大量硝态氮没有得到去除,说明催化铁在中性条件下与NO-3很难发生无机反应. 生物对照组由于外加碳源不足,使得NO-3的还原停留在NO-2阶段,因此有一定量NO-2的积累. 催化铁与生物耦合组NO-3转化率高,排除催化铁与NO-3发生无机反应生成NO-2的可能性,由此推断微生物利用了催化铁体系的电子用于NO-3的还原,生成NO-2; 同时过量的NO-2对其还原有一定的抑制作用. 综合两种作用,催化铁与生物耦合可以促进NO-2的积累. 2.2 催化铁耦合反硝化过程中N2 O释放规律
图 4为初始硝酸盐浓度为50 mg ·L-1、 初始pH 为7.88、 碳氮比为1的催化铁与生物反硝化耦合反应N2 O的产生过程. 在反应开始后,催化铁的存在促进了NO-2的积累,而NO-2对N2 O还原酶有较大的抑制作用,使得N2 O无法继续被还原; 与此同时,催化铁体系不断向溶液中释放Fe2+,Fe2+在pH约为8的环境中几乎完全沉淀,形成亚铁氢氧化物. 而亚铁氢氧化物与NO-2在中性条件下的反应产物主要是N2 O. 在两者的综合作用下N2 O气体在60 min达到最大值. 此时外加碳源已完全耗尽,微生物开始进行内源反硝化,而此时NO-3的浓度已降低至很低,微生物进行N2 O的还原,体系中N2 O含量开始降低,至反应结束时微量的N2 O几乎被完全还原成氮气.
 | 图 4 Fe-50-1,初始pH为7.88催化铁耦合组反硝化过程中N2 O产生规律
Fig. 4 Release of N2 O in denitrification coupled with catalytic iron(Fe-50-1)
|
为对此过程有更精确地描述,表 2为初始硝酸盐浓度为50 mg ·L-1、 初始pH为6.05、 碳氮比为1的催化铁与生物反硝化耦合过程中各个指标的积累量及生成速率.
| 表 2 Fe-50-1,初始pH 6.05催化铁耦合组反硝化过程各指标变化情况 Table 2 Dynamics of NO-3,NO-2,N2 O in denitrification coupled with catalytic iron(initial pH 6.05,Fe-50-1) |
2.3 无机反应与生化反应对N2 O生成的贡献
除生物反应外,Fe(Ⅱ)(化合物中的二价铁,下同)与NO-2发生化学反应也可以产生N2 O[14,15],为了明确催化铁耦合组中无机纯化学与生物作用产生N2 O的比例,开展了初始投加亚硝酸根20 mg ·L-1,碳氮比为1,初始pH 7.5的对比试验.
通过图 5可以看出,无机反应组随着NO-2的消耗,N2 O生成速率逐渐降低,最终浓度(以N计)达到300 μg ·L-1. 同时产物经气相色谱检测,未发现氮气. 主要化学反应为:

对照组中,N2 O释放一直处于较低水平,由此可见,NO-2对生物酶抑制产生的N2 O量远远低于亚铁氧化物与NO-2无机反应产生的N2 O量. 催化铁耦合组中,无机反应与N2 O还原酶抑制作用共同促进产生了N2 O,但生物作用对N2 O的反硝化,降低了N2 O的生成量,经3条曲线对比可知,无机反应的贡献较大. 在图 5中可以发现,在0~180 min内,催化铁组N2 O释放量明显低于无机组,这是由于少量的有机碳源促进了N2 O的还原. 反硝化120 min后,碳源已全部耗尽,亚铁氧化物继续与NO-2发生无机反应产生N2 O,同时N2 O还原酶竞争电子能力较弱[16],其还原也由于电子(有机物提供)的不足而被迫停止,宏观表现为N2 O产生速率增加. 2.4 反应过程中的铁形态
铁为多价态过渡金属,水解过程中较易发生各 种聚合反应,生成具有较长线型结构的多核羟基络
 | 图 5 不同工艺下N2 O产生情况
Fig. 5 Release of N2 O under different conditions
|
| 表 3 反硝化过程中铁离子形态变化 Table 3 Dynamics of iron ion forms during denitrification |
Ksp(FeOH)2=7.9×10-16(25℃),Fe2+开始沉淀的理论pH为6.95,因此,反应体系内亚铁以化合物的形式存在,故酸溶后的Fe2+浓度较高. 随着反应的进行,催化铁体系表面逐渐呈现黑色,氧化层由二价铁与三价铁氧化物混合组成,由于反应活性表面逐渐被覆盖,溶液中铁离子产生速率也慢慢降低.
反应过程中铁离子价态变化为Fe0→Fe2+→Fe3+,可用表中混合液酸溶后Fe2+作为Fe0→Fe2+的参考,同时将30 min内新生成的Fe(Ⅲ)作为Fe2+→Fe3+的参考: 可以看出,在反硝化0~30 min、 30~60 min内,30 min内新生成的三价铁离子多于0 min和30 min时刻亚铁离子量,因此可以判断,Fe0→Fe2+→Fe3+反应速率较快,Fe0被大量腐蚀. Fe(Ⅱ)有短暂积累,但是很快会被NO-2氧化为Fe(Ⅲ),相对应的N2 O产率也处于最高峰. 反硝化进行1h以后,30 min内新生成的三价铁离子少于60 min和90 min时刻亚铁离子量,说明Fe2+→Fe3+反应速率大幅度减慢,体系中所剩NO-2已经不能完全氧化溶液中的Fe(Ⅱ),Fe(Ⅱ)以较慢的速度进行积累. 而随着NO-2与Fe(Ⅱ)反应的减慢,N2 O生成速率小于降解速率,宏观表现为N2 O浓度的下降.
在整个反硝化过程中,体系pH呈现不断上升的趋势,这是由于在较大氧化还原电位的驱使下,铁不断被氧化,H+不断被消耗的结果. 2.5 不同初始pH值对N2 O产生的影响
研究表明,较低的pH会对N2 O还原酶活性产生影响,从而增加N2 O气体的产生(图 6和表 4). 而在催化铁耦合组中,低pH还能促进铁的腐蚀,铁由零价变为二价,新生成的亚铁氢氧化物立即与NO-2反应生成N2 O,因此,pH 6~7组N2 O产生量最大,在90 min达到最大积累量,相比其他两组有一定的延后.
阴极:
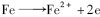
阳极:

 | 图 6 不同pH对催化铁耦合组反硝化过程中N2 O产生影响
Fig. 6 Effect of pH on N2 O emission during denitrification coupled with catalytic iron
|
| 表 4 反应结束时各指标含量 Table 4 Concentrations of total iron ions,NO-2,N2 O after reaction |
2.6 铁离子对反硝化体系ORP的影响
有学者指出[17,18],反硝化段溶解氧的存在会对N2 O的产生有较大的影响. 能否利用催化铁体系高还原能力,消除溶解氧的干扰,由于铁是活泼金属,在水溶液中的腐蚀可以自发进行,主要电极反应式如下.
阳极:
 阴极:
阴极:

因此将催化铁与反硝化耦合可以控制反应体系ORP在一个很低的水平,从而有利于缺氧反硝化的进行,结果如图 7所示.
催化铁耦合组ORP明显低于对照组,越靠近铁刨花骨架,所测ORP越低,铁刨花表面ORP能达到-700 mV,可见铁刨花的存在可以消除反应体系的溶解氧,降低周围氧化还原电位,从而使NO-3成为唯一的有机电子受体被还原.
 | 图 7 反硝化氧化还原电位ORP变化
Fig. 7 Dynamics of ORP during denitrification
|
(1)低碳氮比反硝化过程中,催化铁可以提高硝氮转化率.
(2)低碳氮比催化铁耦合生物反硝化,产生N2 O高于生物对照组,主要源于体系内存在亚铁氧化物与NO-2的无机反应; 催化铁组产生的N2 O低于无机反应组,体系内耦合生物反应减少了N2 O产生.
(3)较低的pH值可以加速催化铁的阳极反应,使NO-2浓度增加,从而增加了N2 O的产生. 催化铁可以降低体系的氧化还原电位,有利于反硝化的还原环境.
| [1] | Kuba T, Van Loosdrecht M C M, Heijnen J J. Phosphorus and nitrogen removal with minimal cod requirement by integration of denitrifying dephosphatation and nitrification in a two-sludge system[J]. Water Research, 1996, 30 (7): 1702-1710. |
| [2] | Hanaki K, Hong Z, Matsuo T. Production of nitrous oxide gas during denitrification of wastewater[J]. Water Science & Technology, 1992, 26 (5-6): 1027-1036. |
| [3] | 彭永臻, 尚会来, 张静蓉, 等. ρ(C)/ρ(N)对污水反硝化过程中N2O产生的影响[J]. 北京工业大学学报, 2010, 36 (4): 517-522. |
| [4] | 尚会来, 彭永臻, 王淑莹, 等. 污水生物脱氮过程中N2O的产生和减量化控制[J]. 中国给水排水, 2008, 24 (16): 104-108. |
| [5] | Beline F, Martinez J, Chadwick D, et al. Factors affecting nitrogen transformations and related nitrous oxide emissions from aerobically treated piggery slurry[J]. Journal of Agricultural Engineering Research, 1999, 73 (3): 235-243. |
| [6] | 马鲁铭. 废水的催化还原处理技术[M]. 北京: 科学出版社, 2008. |
| [7] | Chi J, Zhang S T, Lu X, et al. Chemical reduction of nitrate by metallic iron[J]. Journal of Water Supply: Research and Technology-AQUA, 2004, 53 (1): 37-41. |
| [8] | Huang C P, Wang H W, Chiu P C. Nitrate reduction by metallic iron[J]. Water Research, 1998, 32 (8): 2257-2264. |
| [9] | 王子. 零价铁与生物反硝化耦合脱除硝态氮的研究[D]. 上海: 同济大学, 2012. 44-66. |
| [10] | Zhang Z, Hao Z W, Yang Y P, et al. Reductive denitrification kinetics of nitrite by zero-valent iron[J]. Desalination, 2010, 257 (1-3): 158-162. |
| [11] | Hu H Y, Goto N, Fujie K. Effect of pH on the reduction of nitrite in water by metallic iron[J]. Water Research, 2001, 35 (11): 2789-2793. |
| [12] | Huang Y H, Zhang T C. Nitrite reduction and formation of corrosion coatings in zerovalent iron systems[J]. Chemosphere, 2006, 64 (6): 937-943. |
| [13] | Suzuki T, Moribe M, Oyama Y, et al. Mechanism of nitrate reduction by zero-valent iron: Equilibrium and kinetics studies[J]. Chemical Engineering Journal, 2012, 183: 271-277. |
| [14] | Tai Y L, Dempsey B A. Nitrite reduction with hydrous ferric oxide and Fe(Ⅱ): Stoichiometry, rate, and mechanism[J]. Water Research, 2009, 43 (2): 546-552. |
| [15] | Kampschreur M J, Kleerebezem R, de Vet W W J M, et al. Reduced iron induced nitric oxide and nitrous oxide emission[J]. Water Research, 2011, 45 (18): 5945-5952. |
| [16] | Schalk-Otte S, Seviour R J, Kuenen J G, et al. Nitrous oxide (N2O) production by Alcaligenes faecalis during feast and famine regimes[J]. Water Research, 2000, 34 (7): 2080-2088. |
| [17] | Tallec G, Garnier J, Billen G, et al. Nitrous oxide emissions from denitrifying activated sludge of urban wastewater treatment plants, under anoxia and low oxygenation[J]. Bioresource Technology, 2008, 99 (7): 2200-2209. |
| [18] | Otte S, Grobben N G, Robertson L A, et al. Nitrous oxide production by Alcaligenes faecalis under transient and dynamic aerobic and anaerobic conditions[J]. Applied and Environmental Microbiology, 1996, 62 (7): 2421-2426. |