Distributions of Antibiotic Resistance Genes and Microbial Communities in the Nearshore Area of the Yangtze River Estuary
2006年, Pruden等[1]提出将抗生素抗性基因(antibiotic resistance genes, ARGs)作为一种新型的环境污染物, 世界卫生组织也于2015年明确提出将微生物抗性作为全球健康风险.全世界范围内越来越多的研究人员开始关注ARGs诱发的相关问题.近年来, 抗生素的滥用导致微生物(包括病原菌和非病原菌)对抗生素的抗性逐年增加, 环境中的细菌可以通过基因突变和代系遗传等方式获得抗性基因, 也可以通过转导、接合和转化等方式获得外源存在的抗性基因[2].也有研究表明, 环境中较低浓度的抗生素可以作为微生物种间或者种内的信号分子, 从而调控微生物群落对抗生素进行选择性适应[3].抗生素的存在会加速上述过程的发生, 增加环境中抗生素抗性出现的频率, 对环境安全和人类健康造成潜在威胁.近年来, 我国各大流域都已存在不同程度的ARGs污染[4~7].
海岸带尤其是河口地区, 是连接陆地与海洋的重要过渡区域[8], 接纳了大量来自河流径流和污水排放的污染物, 包括多环芳烃[9]、重金属[10]、抗生素[11]和ARGs[12].含有ARGs的污水最终会通过河口排入海洋环境, 并在人类活动和水文气象等多种因素的耦合影响下[13, 14], 发生一系列复杂的物理-化学-生物作用过程.抗生素抗性基因导致细菌产生耐药性, 并可通过水平转移在细菌间传播扩散[15], 严重威胁生态系统安全和人类健康[16].因此, 厘清河口区域水环境中ARGs和微生物群落的分布特征至关重要.
本文选取长江口近岸地区作为研究区域, 采用荧光定量PCR(quantitative-polymerase chain reaction, qPCR)分析水体和沉积物中共10种ARGs的分布规律, 包括2种磺胺类抗性基因(sul1、sul2)、6种四环素类抗性基因(tetM、tetC、tetX、tetA、tetO、tetQ)以及1种整合子基因intI1和16S rRNA基因, 并采用16S扩增子测序技术分析水环境中微生物群落的分布特征, 揭示长江口近岸地区ARGs与微生物群落间的影响机制, 以期为控制长江口近岸地区水环境中ARGs污染提供数据支持和科学依据, 并为长江口近岸地区ARGs生态风险评价提供参考.
1 材料与方法
1.1 样品采集
本文采集了长江口近岸地区8个采样点的表层水和沉积物样品, 具体采样点分布如图 1所示.自北向南分别是浏河口(1)、石洞口(2)、吴淞口(3)、竹园排污口(4)、三甲港水闸(5)、朝阳农场(6)、大治河(7)和南汇东滩(8).其中浏河口(1)是浏河与长江的交汇地, 吴淞口(3)是黄浦江与长江的交汇地, 大治河(7)是大治河与长江交汇地, 石洞口(2)和竹园(4)位于城市污水处理厂的排污口, 朝阳农场(6)属于潮滩环境, 三甲港水闸(5)位于九段沙湿地国家自然保护区的下游断面, 南汇东滩(8)属于湿地保护区.
有研究表明, 温度对ARGs的传播扩散起着重要的作用[17].因此本次采样时间为2021年7月14日.每个采样点采集表层水(2 L)并装入超纯水洗过的聚乙烯瓶中, 同时采集100 g沉积物(0~5 cm), 并转移至灭菌的塑料袋中.每个采样点采集3个平行样品, 所有样品均用冰袋保存直至运送到实验室.
1.2 DNA提取
1 L水样经0.22 μm孔径的玻璃纤维滤膜过滤并收集滤膜, 沉积物称取5 mg湿样, 然后根据DNA提取试剂盒(Fast DNA Spin Kit for Soil, MP Biomedicals, 美国)说明书提取水样和沉积物样的总DNA.提取的DNA质量和完整性通过1.5%的琼脂糖凝胶电泳进行验证, 并通过超微量分光光度计(NanoDrop2000, 美国)进一步定量DNA的浓度.所有DNA样品存放于-20℃冰箱备用.
1.3 荧光定量PCR
本文通过荧光定量PCR技术(qPCR)定量分析了2类8种目标抗性基因和Ⅰ类整合子基因.本文使用的ARGs片段对应的引物序列、片段大小、退火温度和文献如表 1所示.荧光定量PCR反应体系(20 μL)为:2×Taq Pro Universal SYBR qPCR Master Mix 10 μL, 上下游引物各0.4 μL, DNA模板2 μL, ddH2 O 7.2 μL.基因扩增的热循环反应程序为:95℃预变性30 s; 40个循环(每个循环95℃ 10 s, 退火30 s, 退火温度见表 1); 溶解曲线温度设置为由60℃增加到95℃.
表 1
(Table 1)

表 1 目的基因的qPCR引物信息和反应条件
Table 1 Primers, product lengths, and annealing temperatures of qPCR for target genes
| 目的基因 |
引物序列(5′-3′) |
片段大小/bp |
退火温度/℃ |
文献 |
|
sul1
|
F-CACCGGAAACATCGCTGCA
R-AAGTTCCGCCGCAAGGCT |
158 |
60 |
[18] |
|
sul2
|
F-TCCGGTGGAGGCCGGTATCTGG
R-CGGGAATGCCATCTGCCTTGAG |
191 |
55 |
[19] |
|
tetM
|
F-CCGTTGGGAAGTGGAATGC
R-TCCGAAAATCTGCTGGGGTA |
196 |
57 |
[20] |
|
tetC
|
F-TGCAACTCGTAGGACAGGTG
R-ACCAGTGACGAAGGCTTGAG |
140 |
60 |
[21] |
|
tetX
|
F-AGCCTTACCAATGGGTGTAAA
R-TTCTTACCTTGGACATCCCG |
278 |
60 |
[22] |
|
tetA
|
F-GCTACATCCTGCTTGCCTTC
R-CATAGATCGCCGTGAAGAGG |
210 |
60 |
[20] |
|
tetO
|
F-GATGGCATACAGGCACAGACC
R-GCCCAACCTTTTGCTTCACTA |
172 |
57 |
[20] |
|
tetQ
|
F-AGAATCTGCTGTTTGCCAGTG
R-CGGAGTGTCAATGATATTGCA |
167 |
53 |
[23] |
|
intI1
|
F-CCTCCCGCACGATGATC
R-TCCACGCATCGTCAGGC |
280 |
60 |
[22] |
| 16S rRNA |
341F-CCTACGGGAGGCAGCAG
534R-TTACCGCGGCTGCTGGCAC |
193 |
60 |
[24] |
|
表 1 目的基因的qPCR引物信息和反应条件
Table 1 Primers, product lengths, and annealing temperatures of qPCR for target genes
|
为了生成用于测定提取DNA中每ng基因丰度的qPCR标准曲线, 将目标基因克隆到质粒(E. coli DH5a)中.使用NanoDrop2000测量质粒浓度, 并根据质粒长度和靶序列计算每毫升溶液中靶基因的丰度.通过对携带目的基因的质粒进行10倍的连续梯度稀释, 构建qPCR标准曲线.目的基因的标准曲线和扩增效率如表 2所示.
表 2
(Table 2)
表 2 ARGs定量所需标准曲线和扩增效率1)
Table 2 Standard curve and amplification efficiency of target genes
| 基因名称 |
标准曲线 |
R2 |
扩增效率/% |
|
sul1
|
y=-3.573 8 x+41.395 |
0.998 6 |
91 |
|
sul2
|
y=-3.485 5 x+47.301 |
0.994 0 |
94 |
|
tetM
|
y=-3.142 5 x+37.189 |
0.995 0 |
108 |
|
tetC
|
y=-3.400 5 x+39.997 |
0.998 2 |
97 |
|
tetX
|
y=-3.500 7 x+38.934 |
0.997 8 |
93 |
|
tetA
|
y=-3.374 3 x+37.905 |
0.998 4 |
98 |
|
tetO
|
y=-3.457 3 x+40.483 |
0.999 6 |
95 |
|
tetQ
|
y=-3.847 6 x+43.346 |
0.997 8 |
82 |
|
intI1
|
y=-3.822 x+43.46 |
0.999 2 |
83 |
| 16S rRNA |
y=-3.226 9 x+37.564 |
0.999 3 |
104 |
| 1)
y为qPCR检测的Ct值,
x为计算后的基因拷贝数经lg转化的值 |
|
表 2 ARGs定量所需标准曲线和扩增效率1)
Table 2 Standard curve and amplification efficiency of target genes
|
1.4 16S扩增子测序
为了研究长江口近岸地区水环境的细菌群落多样性, 使用上游引物341F(ACTCCTACGGGAGGC AGCA)和下游引物805R(GGACTACHVGGGTWTC TAAT)对16S rRNA基因V3/V4区域进行扩增.扩增产物送至上海伟寰生物科技有限公司, 采用Illumina NovaSeq PE250平台进行16S扩增子测序.
1.5 数据分析
本文使用Origin软件对所有数据进行汇总分析, 绘制柱状图、相关性热图和聚类分析热图, 用Spearman法进行相关性分析(P < 0.05表示显著相关), 使用R语言包“Hmisc”计算ARGs与微生物群落的相关系数.ARGs丰度变化的主成分分析采用免费的在线数据分析平台Tutools进行分析(https://www.cloudtutu.com), ARGs与微生物群落网络分析图的绘制采用生科云在线数据分析平台(https://www.bioincloud.tech/).
2 长江口近岸地区ARGs的赋存
2.1 ARGs的分布特征
图 2为长江口近岸地区水环境中ARGs相对丰度分布柱状图, W1~W8表示长江口近岸地区1~8号采样点的水样, S1~S8表示1~8号采样点的沉积物样.本文利用16S rRNA基因的丰度对其余目的基因的丰度进行标准化处理, 用来表征单一细菌ARGs的相对丰度(基因拷贝数与16S rRNA拷贝数的比值)[18, 25], 以减少DNA提取效率以及水环境中微生物背景值干扰所造成的误差[26, 27].本文选择的10种ARGs在长江口近岸地区8个采样点的水样和沉积物中的检出率是100%.
如图 2所示, 在水样中, 9种目标抗性基因中tetC的总相对丰度最高, 其相对丰度范围在5.01×10-2~1.76之间, 为长江口近岸地区表层水中的优势基因.在沉积物中, tetM和tetA的总相对丰度处于较高的水平, 相对丰度范围分别在1.51×10-3~1.96×10-1及6.19×10-3~2.19×10-1之间, 为长江口近岸地区沉积物中的优势基因.不同采样点中ARGs的总相对丰度大小分布规律为:W2>W1>W4>W3>W5>W8>W7>W6(水样中); S1>S2>S4>S3>S5>S6>S8>S7(沉积物中).在水样中, W2和W1的ARGs相对丰度显著高于其他采样点; 在沉积物中, S1、S2和S4的ARGs相对丰度显著高于其他采样点.石洞口和竹园采样点位于大型污水处理厂及其邻近水域, 而浏河口采样点位于城市内河与长江下游交汇处.本文分析结果表明ARGs在长江口近岸地区水环境中的富集与污水处理厂的污水排放以及受人类活动影响的城市内河径流有关.该结论与前人研究该区域的结论相似[28, 29].
本文长江口近岸地区水样和沉积物中ARGs的绝对丰度平均值分别为2.32×104~7.66×104 copies ·mL-1和1.41×107~6.59×107 copies ·g-1. Lu等[30]研究了ARGs在渤海和黄海的空间分布, 结果表明在渤海, 目的基因的总绝对丰度范围在2.05×102~7.25×103 copies ·mL-1之间, 而在黄海中, 目的基因的总绝对丰度范围在21.1~8×103 copies ·mL-1之间.在渤海和黄海的河口区域, 目的基因的总绝对丰度范围分别在1.23×104~3.94×105copies ·mL-1和8.91×104~3.67×105copies ·mL-1之间.Chen等[31]对东海海湾沉积物中的27个目的基因进行了研究, 在杭州湾和象山湾河口区域, 目的基因的总绝对丰度范围分别在8.39×106~2.33×107copies ·g-1和1.25×107~2.08×107 copies ·g-1之间.Lu等[25]对辽河口和大辽河口磺胺类抗性基因(sul1、sul2和sul3)进行了表征, 辽河和大辽河口水样ARGs的绝对丰度平均值 <1×103 copies ·mL-1. 杨颖[32]调查了北江流域四环素抗性基因的污染情况, 发现在所检出的四环素抗性基因中, 含量最高的是tetC, 与16S rRNA拷贝数比值范围在8.3×10-2~13.2之间.范长征等[5]分析了湘江长沙河段水体及底泥中4种常见四环素类抗生素的含量特征及其季节变化, 其优势基因tetC和tetA的相对丰度范围在8.2×10-5~7.3×10-3之间.长江口近岸地区ARGs的污染情况在全国处于较高的水平.
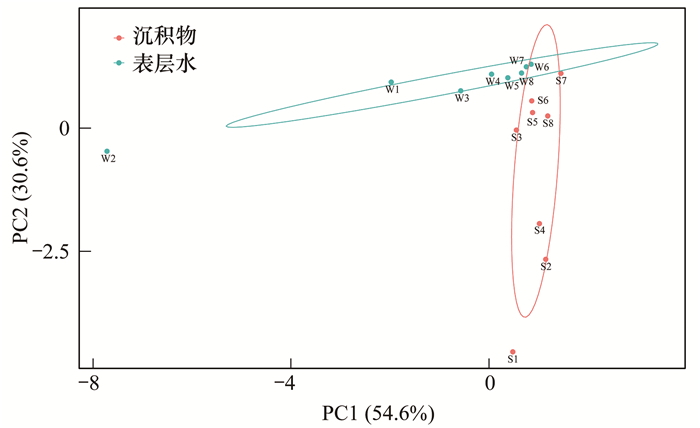
为了更深入地研究长江口近岸地区水环境中ARGs的分布特征以及水体和沉积物ARGs分布的相关性, 本文采用主成分分析法(PCA)[33]对长江口近岸地区8个采样点不同介质中ARGs的变化特征进行聚类分析.如图 3所示, X轴表示第一主成分(PC1), Y轴表示第二主成分(PC2).点与点之间的距离表示差异程度.PC1和PC2总共解释了基因丰度变异的85.2%, 其中, PC1的值为54.6%.可以得出, 表层水样品在PC1上聚集, 沉积物样品在PC2上聚集.说明不同采样点不同介质中的ARGs分别聚类在一起, 而且水样与沉积物样之间没有关联性.只有个别采样点的ARGs分布超出了95%置信椭圆的范围, 分别是水样中的W2和沉积物样中的S1.水样中ARGs的分布变化可能是由于石洞口污水处理厂的污水排放提供了大量的营养物质, 促进了当地的细菌生长.沉积物样中ARGs的分布变化可能是由于浏河口接纳了大量来自城市内河径流和人类活动引起的陆源排放的污染物, 包括重金属、抗生素、抗性细菌等. 浏河口和石洞口样点中ARGs的相对丰度平均值分别为:2.62×10-1和4.50×10-1(水样中), 5.27×10-2和4.32×10-2(沉积物中).这两个样点的ARGs污染水平处于同一数量级, 其差异很小, 而且这两个样点以及吴淞口和竹园排污口的污染水平显著高于其他4个样点, 所以污水处理厂的污水排放和城市内河径流带来的陆源污染物是ARGs污染的主要来源.Proia等[34]通过研究比利时Zenne河中ARGs的分布规律, 发现污水处理厂处理水的排放会增加ARGs的丰度.Guo等[35]研究了ARGs在长江口的赋存特征, 结果表明, 污水影响区域和内河交汇区域会对ARGs的丰度造成较大的影响.
2.2 intI1整合子基因与ARGs的相关性
水平转移是ARGs在环境介质以及微生物中扩散传播的主要途径, 而整合子(integrons)是促进水环境中ARGs水平转移的重要移动基因元件[36~38].
长江口近岸地区8个采样点的intI1的相对丰度从高到低为:W2>W3>W1>W5>W8>W4>W7>W6(水样中); S3>S6>S1>S4>S5>S2>S8>S7(沉积物中).采用Spearman等级相关性检验[39]分析了intI1与其他ARGs的相关关系(如表 3), 说明intI1与多种ARGs都存在显著的相关关系, 尤其是在水样中. sul1基因在水样和沉积物中都与intI1呈显著相关性(P < 0.05), 这可能是由于sul1的基因序列通常与intI1的基因序列重叠所导致, 前人的研究也证实了这一点[40~43].然而, 在沉积物中, 四环素类抗性基因和intI1无显著相关性(P>0.05), 与前人的研究结论不符[44]. Yang等[44]以长江中下游的15个淡水湖泊为研究区域, 对18种不同的ARGs进行检测分析.本研究结果证明四环素类抗性基因tetG与intI1之间存在显著的相关关系(P < 0.05).这说明不同种抗性基因的抗性机制不同, 导致其传播扩散的影响因子不同.虽然intI1是促进水环境中ARGs水平转移的重要移动基因元件, 但自然界中还广泛存在其他可移动遗传元件, 包括整合子、转座子、噬菌体、质粒和基因岛等[45, 46].而且, 其他的环境因素包括抗生素、重金属和多环芳烃等也可能影响表层沉积物中ARGs的传播扩散[47, 48].
表 3
(Table 3)
表 3 表层水和沉积物中intI1与其余ARGs的相关性分析1)
Table 3 Correlation analysis between intI1 and other ARGs in water and sediment samples
| 类型 |
|
sul1
|
sul2
|
tetM
|
tetC
|
tetX
|
tetA
|
tetO
|
tetQ
|
| 表层水 |
r
|
0.881** |
0.048 |
0.881** |
0.881** |
0.786* |
0.786* |
0.786* |
0.690 |
|
P
|
< 0.01 |
0.911 |
< 0.01 |
< 0.01 |
0.021 |
0.021 |
0.021 |
0.058 |
| 沉积物 |
r
|
0.786* |
0.143 |
0.405 |
0.667 |
0.048 |
0.143 |
0.143 |
0.310 |
|
P
|
0.021 |
0.736 |
0.320 |
0.071 |
0.911 |
0.736 |
0.736 |
0.456 |
| 1)
r表示相关系数; *表示显著性差异,
P < 0.05; **表示极显著性差异,
P < 0.01 |
|
表 3 表层水和沉积物中intI1与其余ARGs的相关性分析1)
Table 3 Correlation analysis between intI1 and other ARGs in water and sediment samples
|
3 长江口近岸地区微生物群落分布
3.1 微生物群落多样性分析
本文采用16S扩增子测序技术对长江口近岸地区8个采样点的微生物群落多样性进行分析.细菌群落丰富度指数(Chao1)以及多样性指数(Shannon和Simpson)[49]计算公式如下:

|
(1) |
式中, Sobs为观测到的OTU(操作分类单元)数, n1为只有一条序列的OTU数, n2为只有两条序列的OTU数.
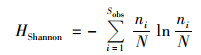
|
(2) |
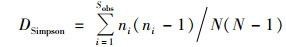
|
(3) |
式中, Sobs为观测到的OTU数, ni为第i个OTU中所含的序列数, N为所有序列数.
如表 4所示, 沉积物的Chao1、Shannon和Simpson指数都高于表层水, 这说明长江口近岸地区沉积物的细菌丰富度和生物多样性要远高于表层水.竹园采样点表层水的Shannon指数在8个采样点中最高(6.80), 沉积物中的Shannon(7.36)指数仅次于吴淞口采样点(7.38), 这可能是因为污水处理厂的污水排放促进了细菌的繁殖, 增加了竹园周围水环境的微生物群落多样性.吴淞口采样点表层水中Simpson指数远远低于其他采样点, 这说明吴淞口水样中的群落物种多样性和均匀度偏低, 其他采样点的Simpson指数都比较接近, 表明物种多样性程度较均匀.
表 4
(Table 4)
表 4 细菌群落丰富度指数和多样性指数
Table 4 Bacterial community richness and diversity index
| 样品 |
类型 |
Chao1指数 |
Shannon指数 |
Simpson指数 |
| 浏河口(1) |
表层水 |
1 554 |
6.18 |
0.993 |
| 沉积物 |
2 431 |
7.06 |
0.998 |
| 石洞口(2) |
表层水 |
928 |
5.42 |
0.985 |
| 沉积物 |
2 521 |
7.31 |
0.999 |
| 吴淞口(3) |
表层水 |
427 |
2.40 |
0.709 |
| 沉积物 |
3 166 |
7.38 |
0.998 |
| 竹园排污口(4) |
表层水 |
1 929 |
6.80 |
0.997 |
| 沉积物 |
2 838 |
7.36 |
0.999 |
| 三甲港水闸(5) |
表层水 |
1 066 |
5.81 |
0.992 |
| 沉积物 |
2 534 |
7.10 |
0.998 |
| 朝阳农场(6) |
表层水 |
818 |
4.96 |
0.969 |
| 沉积物 |
2 272 |
6.70 |
0.995 |
| 大治河(7) |
表层水 |
885 |
5.20 |
0.982 |
| 沉积物 |
2 304 |
7.28 |
0.999 |
| 南汇东滩(8) |
表层水 |
727 |
4.84 |
0.979 |
| 沉积物 |
1 735 |
6.90 |
0.997 |
|
表 4 细菌群落丰富度指数和多样性指数
Table 4 Bacterial community richness and diversity index
|
3.2 微生物群落结构分析
本文对长江口近岸地区8个采样点的微生物群落结构特征进行分析.总体而言, 长江口水环境中的微生物群落由81个门、231个纲、569个目、961个科、1 960个属和4 788个种组成.在门分类水平上, 不同介质中微生物群落的相对丰度如图 4所示.
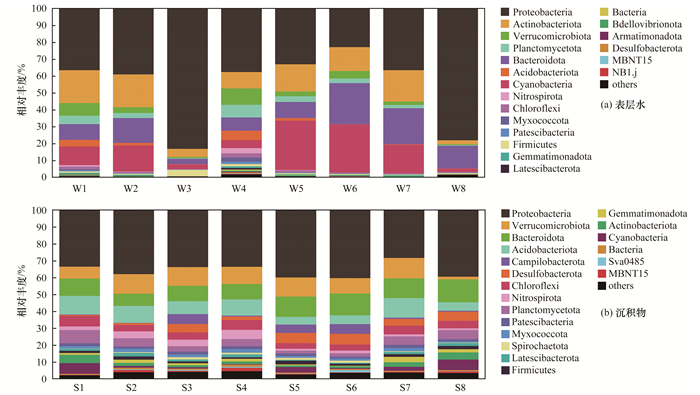
在水样中, 变形菌门(Proteobacteria)、放线菌门(Actinobacteriota)、拟杆菌门(Bacteroidota)和蓝菌门(Cyanobacteria)为主要菌群, 其总相对丰度平均值为85.87%, 其相对丰度平均值占比分别为46%、13.03%、13.00%和13.84%.经过石洞口污水处理厂处理水排放后的下游吴淞口采样点中变形菌门(Proteobacteria)占比显著提高, 占总比例的83.27%, 为绝对优势菌门, 污水处理厂对变形菌门(Proteobacteria)有着显著的影响[50].在沉积物中, 变形菌门(Proteobacteria)、拟杆菌门(Bacteroidota)、疣微菌门(Verrucomicrobiota)和酸杆菌门(Acidobacteriota)为主要菌群, 其总相对丰度平均值为64.06%, 其相对丰度平均值占比分别为36.03%、10.72%、9.23%和8.08%.并且, 变形菌门(Proteobacteria)和拟杆菌门(Bacteroidota)作为长江口水环境中的优势菌群, 可能参与众多物质的生物化学循环过程[51].
本文在属分类水平上对样品及其菌属进行了聚类分析, 并根据各样品中微生物菌属的相对丰度绘制热图, 如图 5所示.在水体中, W1和W5采样点之间的菌属差异较小, 其中相对丰度较高的菌属是Chloroplast、Flavobacterium和Rheinheimera.而W2和W3以及W4和W5采样点之间的菌属差异较大.W2采样点相对丰度较高的是Rheinheimera和Flavobacterium, 分别占总量的31.62%和19.87%; W3采样点的主导菌属为Hydrogenophaga, 其相对丰度占总量的87.17%, 且显著高于其他采样点.在W4采样点相对丰度较高的是Chloroplast和Nitrospira, 分别占总量的14.84%和12.83%; 而W5采样点主导菌属是Chloroplast, 其相对丰度占总量的45.50%. W4和W5采样点之间的微生物菌属存在显著差异, 说明污水处理厂的污水排放对水环境中微生物群落结构变化有较大的影响.有研究发现, 污水处理厂可以直接将微生物释放到环境中[52, 53].在沉积物中, S2和S4采样点微生物分布的相似性较高, 差异较小, 其主导菌属均是硝化螺菌属(Nitrospira), 相对丰度分别占总量的12.46%和11.75%, 这可能是由于污水处理厂的厌氧生物处理工艺增加了水环境中污泥富集硝化螺菌属的风险.硝化螺菌属(Nitrospira)通常是污水处理厂活性污泥体系中的优势菌属[54].
3.3 长江口近岸地区ARGs与微生物相关性分析
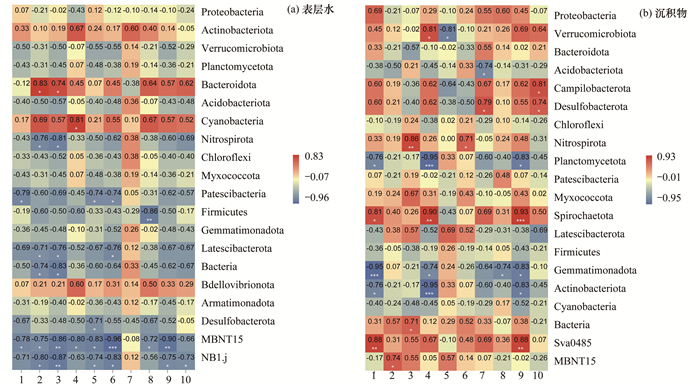
选取相对丰度前20的菌门与ARGs进行Speraman相关性分析, 相关性热图如图 6所示.20种菌门与10种ARGs之间有200种对应关系.在水样中, 菌门与部分ARGs间(29/200)呈显著相关(P < 0.05), 放线菌门(Actinobacteriota)、拟杆菌门(Bacteroidota)和蓝菌门(Cyanobacteria)与大部分ARGs呈正相关.硝化螺旋菌门(Nitrospirota)、髌骨门(Patescibacteria)、厚壁菌门(Firmicutes)、Latescibacterota、Bacteria、Desulfobacterota、MBNT15和NB1.j与ARGs呈显著负相关.在沉积物中, 菌门与部分ARGs间(25/200)呈显著相关(P < 0.05), 变形菌门(Proteobacteria)、Campilobacterota、Desulfobacterota、硝化螺旋菌门(Nitrospirota)、Spirochaetota、Sva0485和MBNT15与多种ARGs呈显著正相关关系.拟杆菌门(Bacteroidota)、浮霉菌门(Planctomycetota)、芽单胞菌门(Gemmatimonadota)和放线菌门(Actinobacteriota)与多种ARGs呈显著负相关关系.当微生物菌门与目的ARGs呈显著正相关关系时, 这些菌门可能是ARGs的潜在宿主.Wang等[55]研究了洪湖中微生物群落和6种目的ARGs之间的相关关系, 发现厚壁菌门(Firmicutes)、变形菌门(Proteobacteria)和芽单胞菌门(Gemmatimonadota)丰度与ARGs丰度呈显著正相关, 表明所属这些门的细菌可能是ARGs的宿主.微生物菌门与目的ARGs呈负相关关系可能意味着该菌门的丰度变化会影响ARGs的丰度和水平转移.Chen等[56]的研究表明, 硫杆菌属(Thiobacillus) 对土霉素具有一定的生物降解作用, 从而可能间接抑制ARGs的水平基因转移, 降低ARGs的相对丰度.
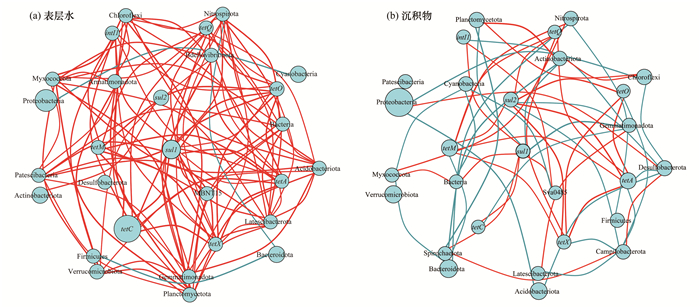
为了分析长江口近岸地区ARGs潜在的宿主信息, 本文采用网络共现图挖掘了ARGs与微生物群落的共现模式(图 7).基于Spearman相关性系数计算结果(r>0.7, P < 0.05), 选取ARGs与相对丰度排名前20的微生物菌门做网络共现分析.结果如图 7所示, 其中的连接线表示节点之间的相关系数大于0.7, 显著性小于0.05的相关关系, 红线表示正相关, 蓝线表示负相关.水样中共检测到5个潜在宿主, 沉积物中共检测到3个潜在宿主.在水样中, 磺胺类抗性基因sul1未检测到宿主信息, sul2只有1个宿主信息(放线菌门), 四环素类抗性基因(tetX、tetA、tetO和tetQ)的潜在宿主较多.在水样中, 硝化螺旋菌门(Nitrospirota)是tetX、tetA、tetO和tetQ共同的潜在宿主.微生物氮循环的频繁发生会产生硝化螺旋菌门(Nitrospirota)[8], 因此, 微生物氮的循环可能会间接影响水体中四环素类抗性基因的分布.在沉积物样品中, 只有sul1、tetC和intI1存在潜在宿主菌门.磺胺类抗性基因sul1的潜在宿主较多(2个), 说明在沉积物中sul1受微生物群落变化的影响较大. sul1的潜在宿主分别为Spirochaetota和Sva0485.硝化螺旋菌门(Nitrospirota)是tetC的潜在宿主.Sva0485是sul1和intI1的共同潜在宿主.由于不同介质中微生物群落结构不同, ARGs的分布和诱导的潜在机制可能有所不同, 从而导致ARGs与微生物群落的相关性以及ARGs的潜在宿主菌存在差异.共同潜在宿主的存在可能会对ARGs的分布特征和水平转移产生影响, 会导致细菌获得多重耐药性, 在一定程度上加速细菌的产生, 生态安全和人类健康可能遭到威胁[57].长江口近岸地区多重耐药菌对ARGs的影响机制值得进一步研究和探索.
4 结论
(1) 2种磺胺类抗性基因(sul1、sul2)、6种四环素类抗性基因(tetM、tetC、tetX、tetA、tetO、tetQ)以及1种整合子基因intI1和16S rRNA基因在长江口近岸地区水环境中均有检出.城市内河径流和污水处理厂的污水排放对长江口近岸地区ARGs的分布有较大的影响.
(2) 在水体中, 整合子基因intI1与7种ARGs(sul1、sul2、tetM、tetC、tetX、tetA、tetO)存在显著正相关关系(P < 0.05); 在沉积物中整合子基因intI1只与一个磺胺类抗性基因sul1存在显著正相关关系.整合子基因intI1可能已经不是沉积物中调控ARGs迁移转化的重要影响因子.
(3) 微生物群落多样性分析上, 长江口近岸地区沉积物中细菌的丰富度和多样性要远高于表层水体.微生物群落结构上, 变形菌门(Proteobacteria)和拟杆菌门(Bacteroidota)是长江口近岸地区水环境中的优势菌门; Chloroplast为水体中的主要菌属, Chloroplast和Nitrospira为沉积物中的主要菌属.微生物群落多样性以及群落结构变化与污水处理厂的污水排放有关.
(4) 在水体中, 硝化螺旋菌门(Nitrospirota)是4种四环素类抗性基因(tetX、tetA、tetO和tetQ)共同的潜在宿主; 在沉积物中, Sva0485是sul1和intI1的共同潜在宿主.微生物群落的丰度变化对长江口近岸地区大部分ARGs的迁移转化有较大影响.
 2023, Vol. 44
2023, Vol. 44