 2021, Vol. 42
2021, Vol. 42


硼酸和磷酸对PMS/Co2+均相催化氧化有机物的影响因素与机制
引用本文
万琪琪, 陈铸昊, 曹瑞华, 王静怡, 文刚. 硼酸和磷酸对PMS/Co2+均相催化氧化有机物的影响因素与机制[J]. 环境科学, 2021, 42(10): 4789-4797.

WAN Qi-qi, CHEN Zhu-hao, CAO Rui-hua, WANG Jing-yi, WEN Gang. Role of Borate and Phosphate Buffers in the Degradation of Organic Compounds in a PMS/Co2+ System: Influencing Factors and Mechanisms[J]. Environmental Science, 2021, 42(10): 4789-4797.
硼酸和磷酸对PMS/Co2+均相催化氧化有机物的影响因素与机制
1. 西安建筑科技大学环境与市政工程学院, 西北水资源与环境生态教育部重点实验室, 西安 710055;
2. 西安建筑科技大学环境与市政工程学院, 陕西省环境工程重点实验室, 西安 710055
2. 西安建筑科技大学环境与市政工程学院, 陕西省环境工程重点实验室, 西安 710055
摘要: 基于过一硫酸盐(PMS)的高级氧化方法被广泛用于有机污染物的降解.本研究以富电子偶氮染料酸性橙7(AO7)为目标有机物,探究了在硼酸(路易斯酸)和磷酸(布朗斯台德酸)两种不同类型的缓冲液中,PMS/Co2+均相催化氧化降解有机物的差异、影响因素及作用机制.PMS/Co2+均相催化氧化体系在磷酸盐缓冲液中降解有机物的k值高于其在硼酸盐缓冲液中,而在磷酸盐缓冲液中的前10 s降解率却低于其在硼酸盐缓冲液中.该体系氧化降解有机物在硼酸和磷酸缓冲液中的差异随着缓冲液、PMS浓度、Co2+浓度和pH的变化而有所不同.PMS/Co2+体系在磷酸盐缓冲液中主要通过SO4-·或·OH氧化降解有机物,在硼酸盐缓冲液中,PMS/Co2+体系主要通过非自由基途径(1O2)降解有机物.该研究将为PMS均相催化中不同类型缓冲液的应用提供参考.
关键词:
PMS/Co2+
硼酸盐
磷酸盐
有机物
自由基
过一硫酸盐
Role of Borate and Phosphate Buffers in the Degradation of Organic Compounds in a PMS/Co2+ System: Influencing Factors and Mechanisms
1. Key Laboratory of Northwest Water Resource, Environment and Ecology, Ministry of Education, School of Municipal and Environmental Engineering, Xi'an University of Architecture and Technology, Xi'an 710055, China;
2. Shaanxi Key Laboratory of Environmental Engineering, School of Municipal and Environmental Engineering, Xi'an University of Architecture and Technology, Xi'an 710055, China
2. Shaanxi Key Laboratory of Environmental Engineering, School of Municipal and Environmental Engineering, Xi'an University of Architecture and Technology, Xi'an 710055, China
Abstract: Peroxymonosulfate(PMS)-based advanced oxidation processes were widely used for the degradation of organic pollutants. Electron-rich azo dye Acid Orange 7(AO7) was selected as the target organic matter in this work. The differences, influencing factors, efficiency, and mechanisms of a PMS/Co2+ homogeneous system in the degradation of organic pollutants with two different buffers of boric acid(Lewis acid) and phosphoric acid(Bronstede acid) were investigated. The k value of AO7 degradation in the PMS/Co2+ homogeneous system with phosphate buffer was greater than that with borate buffer, but the degradation percentage during the first 10 seconds of the reaction was lower in the former case. These differences were affected by buffer concentration, the PMS and Co2+ dosages, and pH. In the phosphate buffer, ·OH or SO4-· contributed to organic degradation in the PMS/Co2+ system, while in the borate buffer, the nonradical pathway(1O2) made a critical contribution to the removal of organics. This study provides a reference for the application of different types of buffers in the homogeneous catalysis of PMS.
Key words:
PMS/Co2+
borate
phosphate
organics
radicals
peroxymonosulfate (PMS)
近年来, 基于过一硫酸盐(KHSO5, PMS)的高级氧化技术引起国内外学者广泛的研究兴趣, 通过一些简单的物理(如光、热和超声等[1~3])或化学(如金属材料、碳材料和某些化学试剂等[4~6])的方法可以催化PMS分解产生一系列的自由基, 如·OH、SO4-·、1O2和O2-·等, 这些具有强氧化性的自由基容易自发地选择性或非选择性地攻击水中难以生物降解的有机污染物, 或将其转化成其他可生物利用的小分子有机物, 或直接矿化成CO2和H2O, 并且尚未发现有害副产物的产生.PMS作为饮用水产品被列入2013年国家卫生计生委综合监督局发布的相关通告中[7], 因此, 基于PMS的高级氧化技术在饮水或污水深度处理的应用前景广阔[8~11].
PMS的均相催化所用到的催化剂主要有两类, 一是金属离子, 如Fe2+、Co2+和Ru3+等; 二是一些化学试剂, 如醌类有机物和卤素离子等[5, 12, 13].PMS的均相催化过程受溶液pH值的影响较大, 还受到水基质的一些成分的影响, 如CO32-、NO3-和NOM等[14].而实验过程中使用的缓冲液成分是否会对PMS均相催化降解有机物的效率产生影响仍需进一步探索.
在生物和化学反应的过程中, 反应速率常常会受到pH值变化的影响, 因此在实验的体系中使用缓冲溶液代替超纯水来维持反应过程的pH稳定.常见的缓冲体系有性质稳定的不挥发性缓冲盐, 如磷酸盐和硼酸盐, 还有稳定性较差的挥发性缓冲盐, 如乙酸盐和碳酸氢盐[15].与磷酸等无机含氧酸不同, 硼酸(H3BO3)作为路易斯酸, 分子结构为平面三角形, 通过夺取水分子电离的OH-而形成四面体结构的B(OH)4-来体现其酸性, 而不是通过去质子化, 身为路易斯碱的B(OH)4-可被归类为亲核试剂[16].硼酸盐溶液常作为缓冲液以维持反应体系pH的稳定, 主要应用缓冲范围为pH 7~10[17].在探索活化或未活化的PMS降解有机污染物的效能时, 许多反应均是在硼酸盐缓冲液中进行[18~21], 但是关于硼酸盐缓冲液和PMS之间相互作用及机制仍有待研究.磷酸盐溶液作为另一种常用缓冲液, 在基于PMS的高级氧化降解有机污染物的研究中, 常被用来控制中性条件[22~25].本研究在考虑硼酸盐缓冲液对PMS的催化氧化降解有机物过程的影响时, 选择磷酸盐缓冲液作为对比, 探究在两种缓冲液中的异同及其原因.
本研究选择了富电子偶氮染料酸性橙7(AO7)为目标有机物, 比较了PMS/Co2+均相体系在硼酸和磷酸缓冲液中氧化降解有机物的效果, 以及不同实验参数条件下该体系在两种缓冲液中降解有机物的效能差异.通过检测PMS/Co2+体系在两种缓冲液中的主要活性物种和反应过程中缓冲液浓度的变化, 阐释了PMS/Co2+体系在硼酸盐和磷酸盐缓冲液中降解有机物过程中产生差异的原因, 以期为PMS/Co2+均相体系在降解有机物中的实际应用提供一定的理论依据.
1 材料与方法 1.1 主要实验试剂与仪器2, 2, 6, 6-四甲基哌啶购自上海阿拉丁生化科技股份有限公司, 磷酸购自天津市天力化学试剂有限公司, 过硫酸氢钾复合盐(Oxone)和5, 5-二甲基-1-吡咯啉-N-氧化物购自西格玛奥德里奇(上海)有限公司, 酸性橙7(AO7)购自上海麦克林生化科技有限公司, 氢氧化钠、硼酸、硫代硫酸钠和硫酸钴均购自天津市科密欧化学试剂有限公司.本实验中所用化学试剂均为分析纯, 用水均采用超纯水机出水.
仪器: 热电阴离子色谱仪(Dionex ICS-1000, 美国)、电子顺磁共振波谱仪(Bruker A300, 德国)和紫外-可见分光光度计[UV2600A, 尤尼柯(上海)仪器有限公司].
1.2 PMS/Co2+催化降解有机物 1.2.1 Co2+/PMS在不同缓冲液中的降解效能配制好0.08 mol·L-1的硼酸溶液, 利用10 mol·L-1的NaOH溶液将其pH调节至所需值7.5, 量取98 mL 0.08 mol·L-1 pH=7.5的硼酸溶液倒入干净的250 mL的烧杯中, 加入1 mL 10 mmol·L-1的AO7, 加入1 mL 100 mg·L-1的Co2+, 接着加入0.1 mL 200 mmol·L-1的PMS引发反应, 在反应时间为10、20、30、60、120、180、300、420和600 s时取样检测AO7的浓度.在磷酸溶液中的反应同上.
1.2.2 影响因素的规律探究PMS浓度的影响: 对于PMS/Co2+体系, 将初始PMS浓度调整为所需值(0.05、0.1、0.2、0.5和1.0 mmol·L-1), 另外的实验参数为0.08 mol·L-1 H3BO3或0.08 mol·L-1 H3PO4、100 μmol·L-1 AO7、1 mg·L-1 Co2+和pH=7.5.
pH的影响: 通过NaOH溶液调节初始的pH为7.0、7.5、8.0和9.0, 另外的实验参数为0.08 mol·L-1 H3BO3或0.08 mol·L-1 H3PO4、0.2 mmol·L-1 PMS、100 μmol·L-1 AO7和1 mg·L-1 Co2+.
硼酸浓度的影响: 将初始硼酸盐浓度调整为所需值(0.02、0.04、0.06、0.08、0.10、0.20、0.30、0.40和0.60 mol·L-1), 另外的实验参数为0.2 mmol·L-1 PMS、100 μmol·L-1 AO7、1 mg·L-1 Co2+和pH=7.5.
磷酸浓度的影响: 将初始磷酸盐浓度调整为所需值(0.02、0.04、0.06、0.08、0.10、0.20和0.30 mol·L-1), 另外的实验参数为0.2 mmol·L-1 PMS、100 μmol·L-1 AO7、1 mg·L-1 Co2+和pH=7.5.
Co2+剂量的影响: 改变Co2+的投加量(0.05、0.1、0.5、1.0和2.0 mg·L-1), 另外的实验参数为0.08 mol·L-1 H3BO3或0.08 mol·L-1 H3PO4、0.2 mmol·L-1 PMS、100 μmol·L-1 AO7和pH=7.5.
1.3 有机物降解机制探究 1.3.1 活性物种的鉴定电子顺磁共振波谱仪(EPR)测定: 在PMS/Co2+体系反应1 min时, 取10 μL的样, 与10 μL的20 mmol·L-1 DMPO或10 mmol·L-1 TEMP的溶液混合均匀, 立刻进行EPR的测定.仪器参数为微波频率9.85 GHz, 扫描宽度10 mT, 中心磁场3 507 mT, 扫描时间为5.24 s, 调制幅度为1 mT.
1.3.2 缓冲液的浓度检测硼酸浓度测定: 利用离子色谱法测定硼酸根离子浓度, 在有机物降解过程中的不同时间点取样.离子色谱仪参数如下: 分析柱与保护柱为AS23柱, 电导抑制器为AERS-4 mm, 抑制电流为23 mA, 柱温为30℃, 淋洗液是量比为3.7 mol∶1.6 mol的碳酸钠与碳酸氢钠混合液, 进样量为25 μL.
磷酸浓度测定: 采用文献[26]中溶解性正磷酸盐的测定方法——钼锑抗分光光度法.
1.4 实验数据分析处理PMS/Co2+体系对AO7的降解过程符合伪一级动力学反应, 根据方程式(1), AO7降解的表观速率常数(k)就是以AO7浓度的自然对数和时间作图求得的斜率, 而表观速率常数的大小可以反映出该降解过程的速度快慢, 通过k值来比较体系的氧化能力.
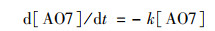
|
(1) |
此外, 当除AO7降解的反应速率常数关于PMS浓度是一阶线性时, 整体反应可以通过二级反应动力学方程式(2)描述:

|
(2) |
式中, k为AO7降解的表观速率常数, [AO7]为AO7的浓度, t为反应时间, [PMS]为PMS的浓度, kapp为二级反应速率常数.
2 结果与讨论 2.1 不同缓冲液对PMS/Co2+均相催化氧化有机物的作用如图 1(a), PMS/Co2+体系在0.08 mol·L-1的磷酸缓冲液中10 min能降解63%的AO7, 而同样时间在相同浓度的硼酸缓冲液中AO7降解率只有50%.

|
图 1 PMS/Co2+体系在不同类型缓冲液中降解AO7 Fig. 1 Degradation of AO7 by the PMS/Co2+ system with different types of buffers |
不论在磷酸缓冲液中还是硼酸缓冲液中, AO7的降解反应均在反应刚开始的瞬间有快速的降解, 因此用两个方法来评价PMS/Co2+体系降解AO7的效能[图 1(b)], 一是前10 s的降解率, 在磷酸和硼酸缓冲液中分别为12%和17%; 二是10 s后通过一级动力学拟合得到的反应速率常数k值, 在磷酸和硼酸缓冲液中分别为0.108 9 min-1和0.070 2 min-1.虽然PMS/Co2+体系在磷酸缓冲液中后阶段的反应速率常数和总的AO7降解率高于在硼酸缓冲液中, 但是在反应开始瞬间(10 s)的降解率却低于在硼酸缓冲液中, 推测是因为硼酸根与Co2+的络合需要一定的作用时间.
2.2 缓冲液浓度的影响规律图 2(a)所示为PMS/Co2+体系在不同浓度的磷酸盐缓冲液中降解AO7的效果, 结果表明, 在pH=7.5时, 磷酸缓冲液浓度从0.02 mol·L-1增加到0.3 mol·L-1, 该体系降解AO7的效率只有微弱的变化, 总降解率的变化范围为53%~66%, 该现象与钴离子催化臭氧除氯过程相似, 几乎不受磷酸浓度的影响[27].根据图 2(b)可以发现, 在磷酸浓度低于0.1 mol·L-1时, 随着磷酸浓度的增加, 前10 s的降解率几乎不变, 而反应速率常数的变化呈现微小的浮动, 在磷酸浓度大于0.1 mol·L-1时, 不论是反应速率常数或是前10 s的降解率均随着磷酸浓度的增加而增加, 在磷酸浓度为0.3 mol·L-1时, 反应速率常数最大为0.135 6 min-1, 前10 s的降解率最大为17%.推测磷酸根离子在该反应体系中充当了桥连的作用, 加快了PMS和Co2+之间的电子转移速度.

|
图 2 PMS/Co2+体系在不同浓度的磷酸盐缓冲液中降解AO7 Fig. 2 Degradation of AO7 by the PMS/Co2+ system with different concentrations of phosphate buffer |
PMS/Co2+体系在不同浓度的硼酸盐缓冲液中降解AO7时, 与在磷酸盐缓冲液中呈现出不同的结果.如图 3(a)所示, PMS/Co2+体系降解AO7的效率受到硼酸缓冲液浓度的影响显著, 总降解率的波动范围为44%~66%.从图 3(b)可看出, 前10 s的降解率随着硼酸浓度的增加而呈现增加的趋势, 而反应速率常数在硼酸浓度为0.02~0.1 mol·L-1时呈降低趋势, 在0.1 mol·L-1硼酸缓冲液中达到最小值0.065 7 min-1, 继续提高硼酸浓度, 反应速率常数逐渐增大, 在0.6 mol·L-1时达到最大值0.261 3 min-1.在没有催化剂的体系中, 高浓度硼酸的存在会减缓有机物的降解速率[28], 因此PMS/Co2+体系中高浓度硼酸的积极作用并非来自硼酸催化PMS, 具体原因仍有待研究.

|
图 3 PMS/Co2+体系在不同浓度的硼酸盐缓冲液中降解AO7及PMS/Co2+体系在不同浓度的硼酸盐和磷酸盐缓冲液中降解AO7的效能对比 Fig. 3 Degradation of AO7 by the PMS/Co2+ system with different concentrations of borate buffer, and a comparison with results obtained with the phosphate buffer |
比较PMS/Co2+体系在磷酸和硼酸缓冲液中降解AO7的效率间的差异[图 3(c)], 发现在不同缓冲液浓度下, 硼酸缓冲液中前10 s降解率始终高于在磷酸缓冲液中, 与此相反, 在硼酸缓冲液中的反应速率常数始终低于在磷酸缓冲液中.值得注意的是, 在缓冲液浓度从0.1 mol·L-1提高到0.3 mol·L-1时, 前10 s降解率和反应速率常数在硼酸和磷酸缓冲液中都呈增加趋势.
2.3 PMS和Co2+剂量的影响 2.3.1 PMS浓度的影响本研究发现, 随着PMS浓度由0.1 mmol·L-1增加至0.5 mmol·L-1, PMS/Co2+体系在磷酸盐缓冲液中降解AO7的效率不断提高[图 4(a)], 前10 s的降解率从7.6%增加到29.2%.在PMS浓度为1.0 mmol·L-1时, 前10 s降解率高达52.5%, 超过一半的AO7在10 s内被氧化[图 4(b)].与此同时, 10 s后AO7的降解速率也随着PMS浓度的增加而加快, 从0.054 6 min-1(0.1mmol·L-1 PMS)显著提高到1.049 4 min-1(1.0 mmol·L-1 PMS), 表明PMS/Co2+体系在磷酸盐缓冲液中的氧化能力受PMS浓度的影响较大, PMS浓度的增加加速了自由基的生成, 促进了AO7的降解.

|
图 4 不同PMS浓度时, PMS/Co2+体系在磷酸盐缓冲液中降解AO7 Fig. 4 Degradation of AO7 by the PMS/Co2+ system at different PMS concentrations in the phosphate buffer |
同样地, PMS/Co2+体系在硼酸盐缓冲液中降解AO7的效果也受到PMS浓度的显著影响, AO7总的降解率随着PMS浓度的提高而增加[图 5(a)].前10 s降解率在PMS浓度从0.1 mmol·L-1增加到0.2 mmol·L-1时几乎不变, 继续提高PMS的浓度到0.5 mmol·L-1、1.0 mmol·L-1, 前10 s降解率分别提高至30.9%、44.6% [图 5(b)]. 10 s后的反应速率常数随PMS浓度的增加成比例地增加(0.289 7 min-1·mmol-1, R2=0.99), 说明PMS/Co2+体系在硼酸盐缓冲液中降解AO7的反应是关于PMS的一级反应.

|
图 5 不同PMS浓度时, PMS/Co2+体系在硼酸盐缓冲液中降解AO7及PMS/Co2+体系在硼酸盐和磷酸盐缓冲液中降解AO7的效能对比 Fig. 5 Degradation of AO7 by the PMS/Co2+ system at different PMS concentrations in the borate buffer, and a comparison with results obtained with the phosphate buffer |
图 5(c)为不同PMS浓度时, PMS/Co2+体系在两种缓冲液中降解AO7的对比, PMS浓度为0.1 mmol·L-1时, PMS/Co2+体系在硼酸盐缓冲液中的前10 s降解率高于在磷酸盐缓冲液中, 随着PMS浓度增加, 该差距逐渐缩小, PMS浓度为1.0 mmol·L-1时, 在硼酸盐缓冲液中前10 s降解率反而低于在磷酸盐缓冲液中, 即加大PMS的浓度可以减少磷酸盐对于初始时Co2+活化PMS的影响.但是在磷酸盐缓冲液中的反应速率常数始终高于在硼酸盐缓冲液中, 说明PMS浓度对PMS/Co2+体系在磷酸盐缓冲液中的氧化能力影响更大.
2.3.2 Co2+剂量的影响在磷酸缓冲液中, Co2+负载量对于PMS/Co2+体系降解AO7的影响非常显著[图 6(a)].随着Co2+剂量从0.05 mg·L-1增加到2.00 mg·L-1, 总降解率从27%提高到71%, 前10 s降解率从5%增至14%[图 6(b)], 反应速率常数也从0.066 3 min-1增至0.141 9 min-1, 说明磷酸根离子并未阻碍钴离子对PMS的催化特性.

|
图 6 不同Co2+剂量时, PMS/Co2+体系在磷酸盐缓冲液中降解AO7 Fig. 6 Degradation of AO7 by the PMS/Co2+ system at different Co2+ dosages in the phosphate buffer |
在硼酸缓冲液中, 随着Co2+剂量从0.05 mg·L-1增加到2.00 mg·L-1, 总降解率从34%提高到61% [图 7(a)], 前10 s降解率从10%提高到23%[图 7(b)], 而反应速率常数先增加后降低, 然后趋于稳定, 在0.1 mg·L-1时达到最大值0.175 5 min-1.因此, 推测硼酸根离子与Co2+间的相互作用与钴离子的浓度密切相关, 进而影响Co2+催化PMS的能力.

|
图 7 不同Co2+剂量时, PMS/Co2+体系在硼酸盐缓冲液中降解AO7及PMS/Co2+体系在硼酸盐和磷酸盐缓冲液中降解AO7的效能对比 Fig. 7 Degradation of AO7 by the PMS/Co2+ system at different Co2+ dosages in the borate buffer; and a comparison with results obtained with the phosphate buffer |
通过比较PMS/Co2+体系在磷酸和硼酸缓冲液中降解AO7的差异[图 7(c)]得出, 不同Co2+剂量时, 在硼酸缓冲液中的前10 s降解率始终高于在磷酸缓冲液中, 这与不同浓度缓冲液条件下的规律相似.在低浓度Co2+时, 在硼酸缓冲液中的反应速率常数高于在磷酸缓冲液中, 而在高浓度的Co2+时, 在硼酸缓冲液中的反应速率常数却低于在磷酸缓冲液中.
2.4 溶液pH对PMS/Co2+体系氧化能力的影响规律在pH为7.0~9.0范围内探究了pH值对于PMS/Co2+体系降解AO7的影响.本研究发现, 在磷酸缓冲液中的降解反应依赖于pH值[图 8(a)], 随着pH值的升高, 总降解率从56%增加到67%, 前10 s降解率从10%增加到22%, 反应速率常数提高了1.65倍[(0.09~0.148 5 min-1, 图 8(b)].

|
图 8 不同pH条件下, PMS/Co2+体系在磷酸盐缓冲液中降解AO7 Fig. 8 Degradation of AO7 by the PMS/Co2+ system under different pH conditions in the phosphate buffer |
如图 9(a)和9(b)所示, pH值对PMS/Co2+体系在硼酸缓冲液中的降解反应的影响规律与在磷酸缓冲液中完全不同, 随着pH从7.0至8.0, 前10 s的降解率和反应速率常数均降低, 在pH=8.0时达到最低值(15%和0.069 9 min-1), 继续提高pH值至10.0, 前10 s的降解率和反应速率常数均上升, 在pH=10.0时达到最大值(54%和0.159 9 min-1).当pH=7.0时, k值为硼酸>磷酸, 在pH=9.0时, 前10 s降解率为磷酸>硼酸[图 9(c)].

|
图 9 不同pH条件下, PMS/Co2+体系在硼酸盐缓冲液中降解AO7及PMS/Co2+体系在硼酸盐和磷酸盐缓冲液中降解AO7的效能对比 Fig. 9 Degradation of AO7 by the PMS/Co2+ system under different pH conditions in the borate buffer, and a comparison with results obtained with the phosphate buffer |
2.5 两种缓冲液对PMS/Co2+均相催化氧化有机物的作用机制 2.5.1 反应活性物种的鉴定
为了更好地了解Co2+在两种缓冲液中催化PMS的机制, 选择了DMPO和TEMP作为自旋捕获剂进行EPR实验, 检测反应过程中产生的活性物种.PMS/Co2+体系分别在两种缓冲液中反应1 min进行取样, 然后立刻加入DMPO进行EPR检测.结果如图 10(a)所示, PMS/Co2+在硼酸盐缓冲液中未发现属于DMPO-·OH和DMPO-SO4-·的特征信号, 而PMS/Co2+在磷酸盐缓冲液中的EPR谱图出现了一组1∶2∶1∶2∶1∶2∶1的窄七重峰, 根据已有报道, 该特征光谱是DMPO的氧化产物DMPO-X[29], 其是由于DMPO被大量的强氧化性物质(如·OH或SO4-·)的过度氧化而形成的.虽然PMS/Co2+在磷酸盐缓冲液中的EPR谱图也没发现属于DMPO-·OH和DMPO-SO4-·的特征信号, 但并不代表该体系中不存在这两种自由基, 因为当DMPO被氧化的速率高于捕获这两种自由基的速率时, 便只会出现DMPO-X的信号, 这说明该体系可能产生大量的·OH或SO4-·[30].

|
图 10 PMS/Co2+体系在硼酸和磷酸缓冲液中的EPR谱图 Fig. 10 EPR spectra of the PMS/Co2+ system in the borate and phosphate buffer |
此外, 利用TEMP捕获非自由基途径的主要活性物种1O2.图 10(b)显示PMS/Co2+在磷酸盐缓冲液中并未发现属于TEMP-1O2的特征信号, 而PMS自身能较慢地分解并产生1O2, 这说明该体系中的PMS主要倾向被Co2+活化产生·OH或SO4-·, 即PMS/Co2+在磷酸盐缓冲液中氧化降解AO7主要是通过自由基途径.PMS/Co2+在硼酸盐缓冲液中的EPR谱图观察到了属于TEMP-1O2的1∶1∶1的三重峰, 说明该体系在硼酸盐缓冲液中10 s后主要生成1O2, 而1O2的氧化能力比·OH或SO4-·弱, 因此PMS/Co2+在硼酸盐缓冲液中10 s后降解AO7的反应速率常数低于在磷酸盐缓冲液中.另外, PMS/Co2+在硼酸盐缓冲液中处理AO7的10 s降解率高于在磷酸盐缓冲液中, 推测是因为前10 s内PMS/Co2+在硼酸盐缓冲液中可能产生了·OH或SO4-·, 之后钴离子与硼酸盐之间的相互作用导致无法持续地生成·OH或SO4-·.
普遍认为二价钴离子活化PMS的机制如方程式(3)~(8)所示, CoOH+的形成会促进PMS的活化, 对于生成SO4-·的方程式(4)相当重要, 生成的三价钴离子也会被HSO5-还原到二价, 而三价钴离子的还原是钴离子价态循环的关键, 是钴离子活化PMS的限速步骤[31].在硼酸盐缓冲液中, B(OH)4-倾向于与Co2+相互作用形成[Co2+—B(OH)4-]+复合物[方程式(9)], 该复合物比较稳定, 抑制了二价钴离子活化PMS产生SO4-·.在磷酸盐缓冲液中, HPO42-则趋向于与Co3+间相互作用而形成[Co3+—HPO42-]+复合物[方程式(10)], 减弱了三价钴离子的还原, 阻碍了1O2的形成.
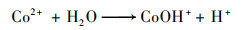
|
(3) |
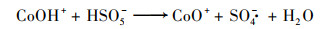
|
(4) |
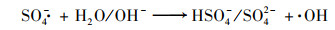
|
(5) |
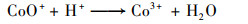
|
(6) |
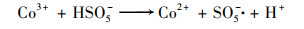
|
(7) |
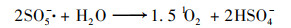
|
(8) |
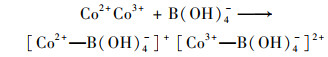
|
(9) |
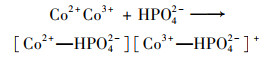
|
(10) |
为了更深入地了解缓冲液对PMS/Co2+体系氧化降解AO7的影响, 检测了整个反应过程中缓冲液浓度的变化.2.5.1节提出了缓冲液的主要离子会与不同价态的钴离子结合形成复合物, 推测在反应完成后缓冲液的浓度应该有所降低.如图 11所示, 磷酸盐和硼酸盐的浓度随着反应的进行基本维持不变, 考虑到所用的缓冲液浓度为0.08 mol·L-1, 该浓度远大于反应体系中钴离子的浓度(16.95 μmol·L-1), 超出了所用的缓冲液浓度测量方法的检测限, 不能据此作出准确的判断, 因此缓冲液对PMS/Co2+体系氧化能力的具体影响机制仍需进一步的探索.

|
图 11 PMS/Co2+体系降解AO7的过程中缓冲液浓度随时间的变化 Fig. 11 Variations in buffer concentration with time during the degradation of AO7 in the PMS/Co2+ system |
3 结论
(1) PMS/Co2+体系在0.08 mol·L-1的磷酸缓冲液中的AO7降解率(63%)高于在硼酸缓冲液中(50%), 在磷酸缓冲液中的k值大于在硼酸缓冲液中, 而前10 s降解率却低于硼酸缓冲液.
(2) 不同缓冲液浓度下, 前10 s降解率是硼酸>磷酸, 反应速率常数则是硼酸 < 磷酸; 磷酸缓冲液中, PMS/Co2+体系的降解效率随着Co2+剂量增加而提高; 在不同Co2+剂量时, 硼酸缓冲液中的前10 s降解率始终高于在磷酸缓冲液中.
(3) 磷酸缓冲液中, PMS/Co2+体系的降解效率随着pH值的增加而提高, 硼酸缓冲液中, PMS/Co2+体系的降解效率随着pH值的增加先降低而后升高.硼酸相对于磷酸更容易提供电子对, Co2+有充足的空轨道, 因此两者倾向于发生络合作用, 降低了Co2+到PMS的电子转移效率.
(4) EPR实验结果表明PMS/Co2+体系在磷酸盐缓冲液中主要通过自由基途径氧化降解有机物, 主要活性物种为·OH或SO4-·, 而PMS/Co2+体系在硼酸盐缓冲液中主要通过非自由基(1O2)途径降解有机物.
参考文献
| [1] | Cai C, Zhang H, Zhong X, et al. Ultrasound enhanced heterogeneous activation of peroxymonosulfate by a bimetallic Fe-Co/SBA-15 catalyst for the degradation of Orange Ⅱ in water[J]. Journal of Hazardous Materials, 2015, 283: 70-79. DOI:10.1016/j.jhazmat.2014.08.053 |
| [2] | Guan Y H, Ma J, Li X C, et al. Influence of pH on the formation of sulfate and hydroxyl radicals in the UV/peroxymonosulfate system[J]. Environmental Science & Technology, 2011, 45(21): 9308-9314. |
| [3] | Ji Y F, Dong C X, Kong D Y, et al. Heat-activated persulfate oxidation of atrazine: Implications for remediation of groundwater contaminated by herbicides[J]. Chemical Engineering Journal, 2015, 263: 45-54. DOI:10.1016/j.cej.2014.10.097 |
| [4] | Anipsitakis G P, Dionysiou D D. Radical generation by the interaction of transition metals with common oxidants[J]. Environmental Science & Technology, 2004, 38(13): 3705-3712. |
| [5] | Fang G D, Gao J, Dionysiou D D, et al. Activation of persulfate by quinones: free radical reactions and implication for the degradation of PCBs[J]. Environmental Science & Technology, 2013, 47(9): 4605-4611. |
| [6] | Sun H Q, Wang Y X, Liu S Z, et al. Facile synthesis of nitrogen doped reduced graphene oxide as a superior metal-free catalyst for oxidation[J]. Chemical Communications, 2013, 49(85): 9914-9916. DOI:10.1039/c3cc43401j |
| [7] | 国家卫生和计划生育委员会通告国卫通[2016]11号[J]. 中华人民共和国国家卫生和计划生育委员会公报, 2016, (8): 8. |
| [8] | Wacławek S, Lutze H V, Grübel K, et al. Chemistry of persulfates in water and wastewater treatment: a review[J]. Chemical Engineering Journal, 2017, 330: 44-62. DOI:10.1016/j.cej.2017.07.132 |
| [9] | Feng M B, Wang Z Y, Dionysiou D D, et al. Metal-mediated oxidation of fluoroquinolone antibiotics in water: a review on kinetics, transformation products, and toxicity assessment[J]. Journal of Hazardous Materials, 2018, 344: 1136-1154. DOI:10.1016/j.jhazmat.2017.08.067 |
| [10] | Wang J L, Wang S Z. Activation of persulfate(PS)and peroxymonosulfate(PMS)and application for the degradation of emerging contaminants[J]. Chemical Engineering Journal, 2018, 334: 1502-1517. DOI:10.1016/j.cej.2017.11.059 |
| [11] | Tsitonaki A, Smets B F, Bjerg P L. Effects of heat-activated persulfate oxidation on soil microorganisms[J]. Water Research, 2008, 42(4-5): 1013-1022. DOI:10.1016/j.watres.2007.09.018 |
| [12] | Wen G, Zhao D, Xu X Q, et al. Inactivation of fungi from four typical genera in groundwater using PMS/Cl- system: efficacy, kinetics and mechanisms[J]. Chemical Engineering Journal, 2019, 357: 567-578. DOI:10.1016/j.cej.2018.09.195 |
| [13] | Li J, Zhou Y, Jiang J, et al. Transformation of phenolic compounds by peroxymonosulfate in the presence of iodide and formation of iodinated aromatic products[J]. Chemical Engineering Journal, 2018, 335: 855-864. DOI:10.1016/j.cej.2017.10.077 |
| [14] | Zhang Y, Wang B J, Hu X N, et al. Non-activated peroxymonosulfate oxidation of p-aminobenzoic acid in the presence of effluent organic matter[J]. Chemical Engineering Journal, 2020, 384. DOI:10.1016/j.cej.2019.123247 |
| [15] |
史倩, 陈军辉, 李鑫, 等. 不同缓冲体系对毛细管电泳法分离15种核苷类化合物效果的比较[J]. 色谱, 2011, 29(6): 481-487. Shi Q, Chen J H, Li X, et al. Comparison of different buffer systems for separation of 15 nucleosides by capillary electrophoresis[J]. Chinese Journal of Chromatography, 2011, 29(6): 481-487. |
| [16] | Peak D, Luther Ⅲ G W, Sparks D L. ATR-FTIR spectroscopic studies of boric acid adsorption on hydrous ferric oxide[J]. Geochimica et Cosmochimica Acta, 2003, 67(14): 2551-2560. DOI:10.1016/S0016-7037(03)00096-6 |
| [17] | Park M, Li Q, Shcheynikov N, et al. NaBC1 is a ubiquitous electrogenic Na+-coupled borate transporter essential for cellular boron homeostasis and cell growth and proliferation[J]. Molecular Cell, 2004, 16(3): 331-341. DOI:10.1016/j.molcel.2004.09.030 |
| [18] | Li J, Jiang J, Zhou Y, et al. Kinetics of oxidation of iodide(I-)and hypoiodous acid(HOI)by peroxymonosulfate(PMS)and formation of iodinated products in the PMS/I-/NOM system[J]. Environmental Science & Technology Letters, 2017, 4(2): 76-82. |
| [19] | Liu J, Zhao Z W, Shao P H, et al. Activation of peroxymonosulfate with magnetic Fe3O4-MnO2 core-shell nanocomposites for 4-chlorophenol degradation[J]. Chemical Engineering Journal, 2015, 262: 854-861. DOI:10.1016/j.cej.2014.10.043 |
| [20] | Zhou Y, Jiang J, Gao Y, et al. Activation of peroxymonosulfate by benzoquinone: a novel nonradical oxidation process[J]. Environmental Science & Technology, 2015, 49(21): 12941-12950. |
| [21] | Zhu S S, Li X J, Kang J, et al. Persulfate activation on crystallographic manganese oxides: mechanism of singlet oxygen evolution for nonradical selective degradation of aqueous contaminants[J]. Environmental Science & Technology, 2019, 53(1): 307-315. |
| [22] | Zheng H, Bao J G, Huang Y, et al. Efficient degradation of atrazine with porous sulfurized Fe2O3 as catalyst for peroxymonosulfate activation[J]. Applied Catalysis B: Environmental, 2019, 259. DOI:10.1016/j.apcatb.2019.118056 |
| [23] | Guan C T, Jiang J, Pang S Y, et al. Nonradical transformation of sulfamethoxazole by carbon nanotube activated peroxydisulfate: kinetics, mechanism and product toxicity[J]. Chemical Engineering Journal, 2019, 378. DOI:10.1016/j.cej.2019.122147 |
| [24] | Nihemaiti M, Permala R R, Croué J P. Reactivity of unactivated peroxymonosulfate with nitrogenous compounds[J]. Water Research, 2020, 169. DOI:10.1016/j.watres.2019.115221 |
| [25] | Feng Y, Wu D L, Deng Y, et al. Sulfate radical-mediated degradation of sulfadiazine by CuFeO2 rhombohedral crystal-catalyzed peroxymonosulfate: synergistic effects and mechanisms[J]. Environmental Science & Technology, 2016, 50(6): 3119-3127. |
| [26] | 国家环境保护总局. 水和废水监测分析方法[M]. 北京: 中国环境科学出版社, 1989. |
| [27] |
张人元, 刘中清, 王琴, 等. 磷酸中臭氧催化氧化除氯[J]. 过程工程学报, 2015, 15(1): 106-110. Zhang R Y, Liu Z Q, Wang Q, et al. Removal of chloride ion from phosphoric acid via ozone catalysis oxidation[J]. The Chinese Journal of Process Engineering, 2015, 15(1): 106-110. |
| [28] | Chen Z H, Wan Q Q, Wen G, et al. Effect of borate buffer on organics degradation with unactivated peroxymonosulfate: influencing factors and mechanisms[J]. Separation and Purification Technology, 2021, 256. DOI:10.1016/j.seppur.2020.117841 |
| [29] | Xu M J, Li J, Yan Y, et al. Catalytic degradation of sulfamethoxazole through peroxymonosulfate activated with expanded graphite loaded CoFe2O4 particles[J]. Chemical Engineering Journal, 2019, 369: 403-413. DOI:10.1016/j.cej.2019.03.075 |
| [30] | Huang J Z, Dai Y F, Singewald K, et al. Effects of MnO2 of different structures on activation of peroxymonosulfate for bisphenol A degradation under acidic conditions[J]. Chemical Engineering Journal, 2019, 370: 906-915. DOI:10.1016/j.cej.2019.03.238 |
| [31] | Chen M T, Zhu L H, Liu S G, et al. Efficient degradation of organic pollutants by low-level Co2+ catalyzed homogeneous activation of peroxymonosulfate[J]. Journal of Hazardous Materials, 2019, 371: 456-462. DOI:10.1016/j.jhazmat.2019.03.002 |