 2021, Vol. 42
2021, Vol. 42

2. 山东省青岛生态环境监测中心, 青岛 266003;
3. 中国海洋大学海洋环境与生态教育部重点实验室, 青岛 266100
 , ZHU Yu-jiao1
, ZHU Yu-jiao1 , MENG He2 , LIU Bing1 , LIU Yu-hong1 , DONG Can1 , YAO Xiao-hong3 , WANG Wen-xing1 , XUE Li-kun1
, MENG He2 , LIU Bing1 , LIU Yu-hong1 , DONG Can1 , YAO Xiao-hong3 , WANG Wen-xing1 , XUE Li-kun1
2. Qingdao Eco-environment Monitoring Center of Shandong Province, Qingdao 266003, China;
3. Key Laboratory of Marine Environmental Science and Ecology, Ministry of Education, Ocean University of China, Qingdao 266100, China
已有的研究表明, 大气颗粒物粒径越小, 其比表面积越大, 越容易吸附有毒有害物质, 也越容易被人体吸入支气管和肺部引起呼吸道类疾病, 甚至可以到达人体的血液系统[1].此外, 超细颗粒物(粒径小于100 nm)可以通过嗅球进入大脑, 引起脑部神经类疾病, 对大脑造成不可逆的损伤[2, 3].新粒子生成(new particle formation, NPF)是大气超细颗粒物的重要来源, 其潜在的健康影响和气候效应受到学界广泛的关注[4, 5].尽管新粒子初始粒径只有1 nm左右, 新粒子粒径增长到大于50 nm可以在较高的过饱和度下(supersaturation, SS, >0.4%)活化成为云凝结核(CCN), 粒径增长到70~80 nm以上则可以在正常过饱和度下(~0.2%)活化成为CCN[6, 7].新粒子增长到100 nm以上则会直接吸收、散射光照辐射[8, 9], 从而影响地气系统的辐射平衡.模式研究表明[10, 11], NPF会使边界层内CCN数浓度增加3%~20%(~0.2% SS)和5%~50%(~1% SS), 进而影响云和气候等相关特性.
海洋气溶胶是全球天然气溶胶的最重要来源[12].在海洋大气中, 浮游植物及生物活动过程中释放的一些挥发性有机前体物对新粒子的生成和增长具有促进作用[13~15].而沿海地区由于同时受到陆源气团和海洋气团的影响, 陆源污染物和海洋生物释放的前体物可能会相互作用, 对新粒子生成及增长的影响较为复杂[6, 16].近年来, 国内外针对沿海地区的NPF事件展开了大量的研究[15, 17~19]. O'Dowd等[14, 20]在爱尔兰西海岸Mace Head频繁观测到NPF事件, 且低潮期藻类释放的碘化物是诱发该地区新粒子事件的重要物种.在我国东海象山湾地区, Yu等[15]和Wan等[21]在海藻养殖期间也观测到了由有机碘化物诱发的新粒子生成事件.在韩国西部沿海地区, Lee等[22]的研究发现NPF事件期间SO2浓度的增加主要受大陆远距离气团传输的影响.Mordas等[23]研究了海、陆气团对NPF事件影响, 发现欧洲Preila站点在海洋气团影响下更易发生NPF事件; 而Babu等[24]通过进一步研究发现, 海陆气团交替传输过程引起的大气湍流运动及边界层的变化对细粒子的暴发及成核过程有促进作用.Kopanakis等[25]基于希腊西北部沿海站点的观测发现, 较高的相对湿度及较低的风速对新粒子成核有促进作用, 而高温和强光照辐射有利于新粒子后续增长过程.
我国东部沿海地区生物活性较强, 沿岸富营养化严重, 海洋生物释放的气态前体物浓度较高[26, 27].青岛处于山东半岛南部, 东、南濒临黄海, 且沿岸为我国黄海浒苔绿潮的最终聚集地, 其独特的地理位置为研究海、陆气团输送的气态前体物对NPF特征的影响提供了有利的条件.本文基于青岛沿岸夏、冬季节观测的大气颗粒物粒径谱数据, 对比新粒子生成和增长特征的季节性差异, 区分海洋生物源和人为源有机物对新粒子生成的影响, 并阐明沿海地区新粒子增长至CCN粒径范围的增长特征及影响因素.
1 材料与方法 1.1 观测时间及地点本研究中夏季观测时间为2019年7月1日~8月19日, 地点位于青岛市即墨区山东大学青岛校区内(东经120°69′, 北纬36°37′), 距离鳌山海水浴场约500 m[如图 1(b)中A点].冬季观测时间为2019年11月10日~2020年1月5日, 地点位于青岛市即墨区青岛海洋科学与技术试点国家实验室(东经120°67′, 北纬36°35′), 距离海岸线约530 [如图 1(b)中B点].冬季和夏季采样地点地处郊区, 东临黄海, 工业排放较少, 周围无明显污染源.两站点相距3 km, 认为受同一天气过程和空气气团影响.

|
(a)SO2排放量分布, 其中SO2排放数据源自更新的亚洲排放清单[28], 单位103 t·(a·网格)-1; (b)观测点地理位置, 源自https://map.baidu.com/, 蓝色圆圈为站点位置:A点为山东大学青岛校区夏季采样点, B点为青岛海洋科学与技术试点国家实验室冬季采样点 图 1 2016年SO2排放量分布及观测点地理位置示意 Fig. 1 Distribution of SO2 emissions in 2016 and location of the sampling site |
此次观测采用美国MSP公司生产的宽范围粒径谱仪(WPS)和德国GRIMM公司生产的电迁移率粒径分析仪(SMPS)对大气颗粒物数浓度及其粒径分布进行实时测量.WPS由微分迁移率分析仪(DMA)、凝结核计数器(CPC)和激光气溶胶分光计(LPS)组成, 其中DMA和CPC分别通过动态电场和工作液的凝聚、凝结液滴的增长对10~500 nm范围内的颗粒物按粒径进行分离和计数, LPS主要通过广角光散射设备实现对350~10 000 nm粒径范围内的颗粒物进行测量[29], 采样时DMA和LPS设定的流量分别为0.3L·min-1和0.7L·min-1, 共分为72个通道进行测量, 时间分辨率为4 min. SMPS由差分迁移率分析仪(L-DMA, 55-U, GRIMM)和超细凝结核粒子计数器(UCPC, 5416, GRIMM)组成.颗粒物首先进入L-DMA中进行筛分以产生单分散气溶胶, 然后进入UCPC测其颗粒物数浓度[30], SMPS设定的采样流量为0.3L·min-1, 时间分辨率为4 min, 共有127个粒径通道.本研究中, WPS放置于采样点A进行夏季观测实验, SMPS放置于采样点B进行冬季观测实验.两台仪器的同步对比观测实验于2020年7月1~7日进行.在同步对比观测实验中, 笔者按照不同粒径段计算了WPS/SMPS的比值(见表 1), 并且将比值按照SMPS的粒径通道进行插值, 作为SMPS校正系数, 以实现两台仪器测量结果的一致性.
|
|
表 1 不同粒径段WPS与SMPS数浓度比值汇总 Table 1 Summary of WPS/SMPS ratios in different particle size ranges |
无机气体污染物如SO2和O3分别使用美国Thermo公司产的43i和49i进行测量, 时间分辨率为1 min.有机气体污染物采用在线VOC监测系统[TT-GCMS, 由英国MARKERS公司生产的前处理设备TT24-7xr及美国Thermo公司生产的Trace 1300(GC)+ISQ(MS)组成]监测, 时间分辨率为1 h. PM2.5采用Thermo 5014i测量, 时间分辨率为1 min, 其化学组分如硝酸盐、硫酸盐和铵盐等, 使用在线离子色谱(AIM-URG, Thermo)在冬季进行测量, 时间分辨率为1 h.辐射数据使用美国国家环境预测中心(NCEP, https://rda.ucar.edu/datasets/ds083.3/)再分析数据集, 该数据集提供了某一时刻值及后3h均值的下行短波辐射.本文使用正午11:00(UTC+8)的辐射值进行计算.
1.3 相关参数计算方法新粒子生成事件的判别标准参照胡敏等[31]和Kulmala等[32]提出的方法:①颗粒物数浓度谱有新的模态产生; ②该模态从 < 25 nm粒径范围内开始出现; ③新的模态可以持续一段时间(大于1 h), 且后续有增长的趋势.
新粒子表观生成速率(FR, cm-3·s-1)参照Kulmala等[32]总结的方法进行计算, 其公式见式(1):
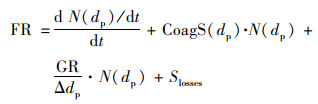
|
(1) |
式中, N(dp)代表核模态颗粒物数浓度, 本文计算时核模态粒径选在10~25 nm的范围内; d N(dp)/dt代表核模态颗粒物数浓度随时间的变化; CoagS(dp)代表碰并汇, 其与N(dp)的乘积表示颗粒物之间碰并过程中引起的损失通量; GR·N(dp)/Δdp表示颗粒物增长到粒径超过25 nm产生的损失; Slosses代表其它过程引起颗粒物数浓度的变化, 一般可忽略不计.由于本文使用仪器的起始粒径较大, 因此, 对于新粒子生成事件的判定时间有所延迟, 实际新粒子生成时间比仪器观测到时间提前约1~3 h.
气溶胶粒径分布依据Whitby等[33]和Zhu等[34]研究中的多正态函数分布公式进行拟合[式(2)]:
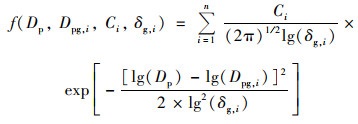
|
(2) |
式中, Dp代表颗粒物粒径, i代表第i个正态分布模态, n代表正态分布模态的个数; 在第i个正态分布模态中, Ci代表颗粒物数浓度, δg, i2代表几何差异系数, Dpg, i代表几何中值粒径;
表观增长速率(growth rate, GR)是指核模态颗粒物的几何中值粒径在一定时间内的变化速率[34], 单位为nm·h-1, 其计算公式见式(3):
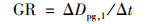
|
(3) |
式中, Dpg, 1代表新粒子模式的几何中值直径.
凝结汇(coagulation sink, CS)表征大气中的可凝结蒸汽凝结在预先存在的颗粒物表面引起的损失[35], 其单位为s-1, 计算公式见式(4):
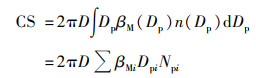
|
(4) |
式中, D代表冷凝蒸汽的扩散系数; βM代表过渡校正因子; Dpi表示第i个通道内颗粒物的粒径; Npi代表第i个通道内颗粒物的数浓度.
此外, 本文所用后向气流轨迹采用混合型单粒子拉格朗日综合轨迹模式(HYbrid single-particle lagrangian integrated trajectory, HYSPLIT)模拟, 模拟时间为48 h, 高度为500 m.
2 结果与讨论 2.1 NPF事件发生频率及颗粒物粒径分布特征概述根据1.3节的判别标准, 观测期间夏季和冬季NPF事件发生天数分别为7 d和15 d, 各占有效观测天数的18%和27%, 冬季较夏季更易发生NPF事件.此频率略低于我国内陆地区NPF事件的发生频率(30%~40%)[4].根据当天是否有NPF事件发生, 将观测天区分为NPF事件天和非NPF事件天, 其粒径分布如图 2所示.

|
图 2 夏季和冬季颗粒物数浓度粒径分布特征 Fig. 2 Average particle number size distribution in summer and winter |
观测期间, 夏季颗粒物总数浓度平均为(1.6±1.2)×104个·cm-3, 略高于冬季[(1.3±0.7)×104个·cm-3].这与我国内陆城市的观测结果不同, 如在北京[36]和济南[37]等地, 冬季颗粒物总数浓度均值为夏季的1.6~1.7倍.如图 2(a), 夏季非NPF天中, 大气颗粒物总数浓度为(1.3±2.5)×104个·cm-3, 颗粒物中值粒径为55nm.新粒子生成时(除7月20日), 大气颗粒物总数浓度升高至(2.6±4.9)×104个·cm-3, 约为非NPF天的1.9倍, 颗粒物中值粒径峰值约为50 nm.值得注意的是, 7月20日[图 2(b)]发生的新粒子生成事件中, 颗粒物总数浓度升高至(5.5±4.2)×104个·cm-3, 其中核模态(10~25 nm)颗粒物数浓度为(2.6±4.1)×104个·cm-3, 该日表观生成速率为125.9 cm-3·s-1, 约为其他天的9~268倍(见表 2), 与Yu等[15]在我国东海象山湾观测到的由碘化物引发的新粒子生成事件特征相似, 因此, 笔者推断7月20日的新粒子生成可能与碘化物促进成核过程有关, 但此天不作为本文的主要内容进行阐述.
|
|
表 2 2019年观测点和已有研究中NPF参数统计1) Table 2 Characterization of NPF events in the observation site and previous studies |
在冬季, 非NPF天中大气颗粒物总数浓度为(1.3±0.7)×104个·cm-3, 颗粒物峰值出现在45 nm左右, 当有NPF事件发生时, 颗粒物总数浓度并没有明显增加, 约为(1.3±0.6)×104个·cm-3. 57 nm以下颗粒物数浓度显著升高, 大于57 nm的颗粒物则数浓度降低, 说明冬季NPF易发生在空气较为清洁、凝结汇较低的环境条件下.NPF天大气颗粒物峰值为35 nm左右, 其粒径小于夏季, 说明新粒子粒径的增长在冬季小于夏季.
2.2 气象参数和气态前体物对夏、冬季节新粒子生成事件的影响图 3是夏、冬两季NPF天和非NPF天气象参数及气态前体物对比.从图 3(a)中可以看出, 温度(T)对夏季和冬季是否发生NPF事件无明显影响.NPF事件易发生在相对湿度(RH)较低的晴天[图 3(b)], 这是因为RH高时通常太阳辐射相对较弱[图 3(c)], 此时VOC的臭氧分解反应及硫酸气体生成反应受限制, 会抑制成核所必需的可凝结蒸汽的形成[40, 41].因此, RH与NPF事件存在表面负相关性.

|
图 3 夏季和冬季气象和气体参数统计 Fig. 3 Meteorological and gaseous precursors in summer and winter |
夏季由于温度较高、太阳辐射较强, 大气氧化性明显高于冬季[图 3(c)和3(e)], 但是相同季节中NPF和非NPF天O3浓度并无显著差别.夏季SO2浓度较低, 其质量分数约为1.7×10-9, 并在NPF天略高于非NPF天.冬季受供暖等因素影响导致SO2浓度明显高于夏季, 且在非NPF天高于NPF天, 这是由于冬季高SO2浓度通常伴随着高浓度大气颗粒物, 即高凝结汇[图 3(f)], 抑制NPF的发生.
污染物浓度日变化显示(图 4), 夏季SO2在NPF天和非NPF天的08:00左右上升到最大值, 且NPF天的峰值浓度高于非NPF天.而冬季NPF天的SO2浓度低于非NPF天.O3日间混合比在两季的NPF天和非NPF天没有显著差异, 说明在相同季节中, 观测点NPF事件的发生基本不受其浓度的制约.但PM2.5浓度在NPF天整体低于非NPF天, 符合低凝结汇利于NPF发生的共识.

|
图 4 夏季和冬季SO2、O3和PM2.5浓度日变化 Fig. 4 Average diurnal variations in SO2, O3, and PM2.5 concentrations in summer and winter |
NPF事件的发生往往受气团来源的影响[31, 42].青岛属温带季风气候, 夏季以海洋气团影响为主, 冬季以陆源气团影响为主.如图 5所示, 海洋源气团在夏季占51.4%, 而冬季只有5.6%, 且冬季后向气流轨迹传输距离普遍大于夏季, 表明冬季气团移动速度相对较快.在海洋气团影响下, 仅有夏季两天(2019-07-20和2019-08-02)发生NPF事件.进一步研究表明, 夏季海洋气团中RH平均为67.9%, 高于陆源气团(57.3%), 因此, 海洋气团中低NPF发生频率可能是受高RH的影响.海洋大气中缺乏气态前体物SO2也是可能的因素.尽管夏季观测中仍发现一定浓度的SO2, 这些SO2可能来自局地陆源而非海洋源.

|
图 5 观测点夏季和冬季48 h后向气团轨迹 Fig. 5 The 48 h air mass backward trajectories at the sampling site in summer and winter |
以往研究表明, 生物源有机物(BVOCs)和人为源有机物(AVOCs)可以参与大气中的成核过程, 但其作用机制仍存在很大的不确定性[43, 44].本研究中选取了部分AVOCs如苯、甲苯、乙炔, 以及BVOCs如异戊二烯, 分析其与NPF之间的关系.如图 6所示, 这几种物质与FR的相关性存在季节性差异.在夏季, 苯(r=-0.52)、甲苯(r=-0.69)和乙炔(r=-0.81)这3种AVOC与FR呈负相关, 而异戊二烯与FR呈正相关, 且相关性较强(r=0.98), 说明夏季BVOCs对新粒子生成可能有直接或间接促进作用, AVOCs对新粒子生成起抑制作用, 但原因有待研究.另外一种可能, 存在的负相关是由于其它因素引起, 二者没有必然联系.而在冬季, 由于生物源排放量明显降低, 异戊二烯与FR呈现非常弱的相关性(r=-0.06), 而该季节苯、甲苯、乙炔与FR呈正相关, r在0.40~0.48之间, 暗示冬季AVOCs对新粒子生成可能起促进作用.人为源有机物的排放通常伴随颗粒物浓度的升高, 因此, 观测期间AVOCs与FR的相关关系也可能是气态源和凝结汇相互竞争结果.但由于此次研究周期较短, 可参考数据量少, 其相关性可能存在一定的偶然性, 尚待深入研究.

|
图 6 苯、甲苯、乙炔及异戊二烯与FR的关系 Fig. 6 Relationship between benzene, toluene, acetylene, and isoprene with FR |
对于GR与上述4种物质间的相关性, 笔者发现除冬季异戊二烯与GR相关性(r=0.5)相对较强之外, 其他物质与GR相关性均小于0.3, 说明新粒子增长过程中, 异戊二烯氧化产物可能有重要的贡献.此外, 有研究发现只有在特定温度及粒径范围内单萜类VOCs与GR之间才具有相关性[45].
2.4 NPF事件的潜在气候效应探讨新粒子的气候效应取决于其粒径的增长.在一定过饱和度下, 粒径越大, 颗粒物越易活化为CCN, 其气候效应越为显著[46]. 本研究中根据新粒子几何中值粒径能达到的最大值(Dpg, max)将NPF事件分为两类:Dpg, max < 50 nm作为第一类NPF事件[以2019-11-21为例, 见图 7(a)], 此类事件几乎不具有气候效应; Dpg, max>50 nm为第二类NPF事件[以2019-08-17及2019-12-02为例, 见图 7(b)和7(c)], 此类事件具有潜在气候效应.观测期间NPF事件的相关参数数值及分类结果见表 2.

|
图 7 青岛新粒子生成事件等值线图及风速风向时间序列 Fig. 7 Contour plot of NPF events and time series of wind in Qingdao |
在观测期间共22 d NPF事件中, 有8 d NPF事件归为第一类, 其新粒子成核、增长到最终消失持续时间平均为4 h.在此类NPF事件持续过程中, 风速(WS)和风向(WD)没有明显的改变[如图 7(a)].共有14 d NPF事件归为第二类, 其持续时间平均为48 h, 为第一类的12倍.此类NPF事件通常持续至夜间或延续2~3 d, 受沿海地区海陆风影响, 其风速和风向在夜间通常会发生变化[如图 7(b)和7(c)].
对于第二类NPF事件, 新粒子的增长过程在夏季和冬季有所不同.如图 7(b), 在夏季, 新粒子生成后可持续增长至CCN粒径, 该过程可能与夏季较强的光化学反应有关.而在冬季发生的10次第二类NPF事件中, 有5d经历了两阶段增长:即白天和夜间出现两次增长过程.这与Zhu等[34]和Man等[47]观测结果类似.以2019年冬季12月2日[如图 7(c)]为例, 10:00开始观测到新粒子生成事件, 新粒子粒径从10 nm增长至30 nm, 增长速率为2.9 nm·h-1.在18:00开始出现第二阶段增长, 至22:00新粒子粒径从30 nm增长至90 nm, 增长速率为15.3 nm·h-1.在第二阶段增长中, 风速小于2 m·s-1, 说明局地污染物的累积可能对颗粒物粒径增长起作用.在冬季5 d的两阶段增长例子中, 白天第一阶段增长使新粒子粒径增长至30~45 nm, 傍晚或夜间的第二阶段增长可以使新粒子粒径从30~45 nm增长到55~90 nm.因此, 夏季光化学过程可以使新粒子增长至CCN粒径, 而冬季往往需要经历夜间增长过程才可使新粒子增长至CCN粒径范围.
2.5 离子组分对新粒子增长的影响对于冬季NPF天, 笔者对比离子组分对粒径增长至CCN粒径范围的影响.图 8所示为第一类NPF事件天、第二类NPF事件天中Dpg≤50 nm时间段和Dpg>50 nm时间段各离子组分的质量浓度及占比.如图 8(a)所示, 第一类NPF天和第二类NPF天中Dpg增长至小于50 nm时, NH4+、SO42-、NO3-和Cl-平均浓度和占比差异不大.但随着第二类NPF事件中新粒子粒径的持续增长, 在粒径超过50 nm之后, 硝酸盐、硫酸盐和铵盐的浓度明显增加.其中, 硝酸盐浓度涨幅最大, 其浓度分别是第一类NPF事件和第二类(Dpg≤50 nm)的7~8倍, 占比从第一阶段增长时的6.7%增长至17.6%[图 8(b)].而硫酸盐占比略有下降, 说明其在NPF事件后续增长中的作用逐渐减弱.因此, 冬季沿海地区中颗粒态硝酸盐的生成对于新粒子的第二阶段增长起重要作用, 且是新粒子增长到CCN粒径范围的关键.

|
图 8 PM2.5化学组分浓度及占比 Fig. 8 Chemical concentrations and their proportions in PM2.5 |
(1) 在共计93 d的观测实验中, 夏、冬两季NPF事件发生天数分别为7 d和15 d, 各占有效天数的18%和27%, 青岛沿海地区冬季更易发生NPF事件.观测站点夏季和冬季颗粒物总数浓度均值分别为(1.6±1.2)×104个·cm-3和(1.3±0.7)×104个·cm-3, 夏季略高于冬季.夏季NPF事件的发生可以使大气颗粒物数浓度增加1~4倍, 但冬季NPF事件对颗粒物总数浓度的影响不大.
(2) 结合气象条件及气态前体物研究发现, 该地区在陆源气团、低相对湿度和低凝结汇条件下, 更易发生NPF事件; 沿海地区夏季氧化性高, 生物活性强, 其释放的BVOC可能对新粒子的生成有促进作用, 而AVOC与FR呈明显的负相关, 其对新粒子生成可能起抑制作用, 但也可能存在其它解释; 冬季与之相反, NPF事件几乎不受BVOC的影响, 而AVOC与FR呈正相关, 相关系数在0.40~0.48之间, 暗示其对新粒子的生成可能存在促进作用.但由于研究数据量较少, NPF相关参数与VOCs间的相关性可能存在一定的偶然性, 需做进一步研究论证.
(3) 对于具有潜在气候效应的NPF事件, 夏季光化学过程可使新粒子直接增长到CCN粒径范围; 而冬季, 新粒子大多需经历白天和夜间两阶段增长, 且夜间的第二阶段增长可以使新粒子增长到CCN粒径范围.其中, 硝酸盐的生成是驱动该过程发生的关键性因素.
| [1] | Rao S, Chirkov V, Dentener F, et al. Environmental modeling and methods for estimation of the global health impacts of air pollution[J]. Environmental Modeling & Assessment, 2012, 17(6): 613-622. DOI:10.1007/s10666-012-9317-3 |
| [2] | Maher B A, Ahmed I A M, Karloukovski V, et al. Magnetite pollution nanoparticles in the human brain[J]. Proceedings of the National Academy of Sciences of the United States of America, 2016, 113(39): 10797-10801. DOI:10.1073/pnas.1605941113 |
| [3] | Collingwood J, Dobson J. Mapping and characterization of iron compounds in Alzheimer's tissue[J]. Journal of Alzheimer's Disease, 2006, 10(2-3): 215-222. DOI:10.3233/JAD-2006-102-308 |
| [4] | Chu B W, Kerminen V M, Bianchi F, et al. Atmospheric new particle formation in China[J]. Atmospheric Chemistry and Physics, 2019, 19(1): 115-138. DOI:10.5194/acp-19-115-2019 |
| [5] | Lee S H, Gordon H, Yu H, et al. New particle formation in the atmosphere: from molecular clusters to global climate[J]. Journal of Geophysical Research: Atmospheres, 2019, 124(13): 7098-7146. DOI:10.1029/2018JD029356 |
| [6] | Zhu Y J, Li K, Shen Y J, et al. New particle formation in the marine atmosphere during seven cruise campaigns[J]. Atmospheric Chemistry and Physics, 2019, 19(1): 89-113. DOI:10.5194/acp-19-89-2019 |
| [7] | Petters M D, Kreidenweis S M. A single parameter representation of hygroscopic growth and cloud condensation nucleus activity-part 3: including surfactant partitioning[J]. Atmospheric Chemistry and Physics, 2013, 13(2): 1081-1091. DOI:10.5194/acp-13-1081-2013 |
| [8] | Dusek U, Frank G P, Hildebrandt L, et al. Size matters more than chemistry for cloud-nucleating ability of aerosol particles[J]. Science, 2006, 312(5778): 1375-1378. DOI:10.1126/science.1125261 |
| [9] | Clarke A D, Davis D, Kapustin V N, et al. Particle nucleation in the tropical boundary layer and its coupling to marine sulfur sources[J]. Science, 1998, 282(5386): 89-92. DOI:10.1126/science.282.5386.89 |
| [10] | Spracklen D V, Carslaw K S, Kulmala M, et al. Contribution of particle formation to global cloud condensation nuclei concentrations[J]. Geophysical Research Letters, 2008, 35(6). DOI:10.1029/2007GL033038 |
| [11] | Yu F Q, Luo G, Nair A A, et al. Wintertime new particle formation and its contribution to cloud condensation nuclei in the Northeastern United States[J]. Atmospheric Chemistry and Physics, 2020, 20(4): 2591-2601. DOI:10.5194/acp-20-2591-2020 |
| [12] | O'Dowd C D, de Leeuw G. Marine aerosol production: a review of the current knowledge[J]. Philosophical Transactions of the Royal Society A: Mathematical, Physical and Engineering Sciences, 2007, 365(1856): 1753-1774. DOI:10.1098/rsta.2007.2043 |
| [13] | Lana A, Simó R, Vallina S M, et al. Potential for a biogenic influence on cloud microphysics over the ocean: a correlation study with satellite-derived data[J]. Atmospheric Chemistry and Physics, 2012, 12(17): 7977-7993. DOI:10.5194/acp-12-7977-2012 |
| [14] | O'Dowd C D, Jimenez J L, Bahreini R, et al. Marine aerosol formation from biogenic iodine emissions[J]. Nature, 2002, 417(6889): 632-636. DOI:10.1038/nature00775 |
| [15] | Yu H, Ren L L, Huang X P, et al. Iodine speciation and size distribution in ambient aerosols at a coastal new particle formation hotspot in China[J]. Atmospheric Chemistry and Physics, 2019, 19(6): 4025-4039. DOI:10.5194/acp-19-4025-2019 |
| [16] | Kerminen V M, Chen X M, Vakkari V, et al. Atmospheric new particle formation and growth: review of field observations[J]. Environmental Research Letters, 2018, 13(10). DOI:10.1088/1748-9326/aadf3c |
| [17] | Flanagan R J, Geever M, O'Dowd C D. Direct Measurements of new-particle fluxes in the coastal environment[J]. Environmental Chemistry, 2005, 2(4): 256-259. DOI:10.1071/EN05069 |
| [18] | Saiz-Lopez A, Plane J M C, Baker A R, et al. Atmospheric chemistry of iodine[J]. Chemical Reviews, 2012, 112(3): 1773-1804. DOI:10.1021/cr200029u |
| [19] | Sorribas M, Adame J A, Olmo F J, et al. A long-term study of new particle formation in a coastal environment: meteorology, gas phase and solar radiation implications[J]. Science of the Total Environment, 2015, 511: 723-737. DOI:10.1016/j.scitotenv.2014.12.011 |
| [20] | O'Dowd C, McFiggans G, Creasey D J, et al. On the photochemical production of new particles in the coastal boundary layer[J]. Geophysical Research Letters, 1999, 26(12): 1707-1710. DOI:10.1029/1999GL900335 |
| [21] | Wan Y B, Huang X P, Jiang B, et al. Probing key organic substances driving new particle growth initiated by iodine nucleation in coastal atmosphere[J]. Atmospheric Chemistry and Physics, 2020, 20(16): 9821-9835. DOI:10.5194/acp-20-9821-2020 |
| [22] | Lee S H, Young L H, Benson D R, et al. Observations of nighttime new particle formation in the troposphere[J]. Journal of Geophysical Research, 2008, 113(D10). DOI:10.1029/2007JD009351 |
| [23] | Mordas G, Plauškaitè K, Prokop Dč iuk N, et al. Observation of new particle formation on Curonian Spit located between continental Europe and Scandinavia[J]. Journal of Aerosol Science, 2016, 97: 38-55. DOI:10.1016/j.jaerosci.2016.03.002 |
| [24] | Babu S S, Kompalli S K, Moorthy K K. Aerosol number size distributions over a coastal semi urban location: Seasonal changes and ultrafine particle bursts[J]. Science of the Total Environment, 2016, 563-564: 351-365. DOI:10.1016/j.scitotenv.2016.03.246 |
| [25] | Kopanakis I, Chatoutsidou S E, Glytsos T, et al. Impact from local sources and variability of fine particle number concentration in a coastal sub-urban site[J]. Atmospheric Research, 2018, 213: 136-148. DOI:10.1016/j.atmosres.2018.06.002 |
| [26] | Guo T, Li K, Zhu Y, et al. Concentration and size distribution of particulate oxalate in marine and coastal atmospheres- Implication for the increased importance of oxalate in nanometer atmospheric particles[J]. Atmospheric Environment, 2016, 142: 19-31. DOI:10.1016/j.atmosenv.2016.07.026 |
| [27] | Xie H, Feng L M, Hu Q J, et al. Concentration and size distribution of water-extracted dimethylaminium and trimethylaminium in atmospheric particles during nine campaigns- Implications for sources, phase states and formation pathways[J]. Science of the Total Environment, 2018, 631-632: 130-141. DOI:10.1016/j.scitotenv.2018.02.303 |
| [28] | Zhang Q, Streets D G, Carmichael G R, et al. Asian emissions in 2006 for the NASA INTEX-B mission[J]. Atmospheric Chemistry and Physics, 2009, 9(14): 5131-5153. DOI:10.5194/acp-9-5131-2009 |
| [29] |
王忠训, 曹大勇, 张玉玲, 等. 大气颗粒物数浓度及粒径分布测量技术: 宽范围颗粒物分光计[J]. 现代科学仪器, 2008, 18(1): 112-116. Wang Z X, Cao D Y, Zhang Y L, et al. Survey Technology of atmospheric particle number concentration and diameter distribution: wide-range particle spectrometer[J]. Modern Scientific Instruments, 2008, 18(1): 112-116. |
| [30] |
王飞, 朱彬, 康汉清, 等. APS-SMPS-WPS对南京夏季气溶胶数浓度的对比观测[J]. 中国环境科学, 2011, 31(9): 1416-1423. Wang F, Zhu B, Kang H Q, et al. Comparative observations on summer aerosol number concentration measured by APS-SMPS-WPS in Nanjing area[J]. China Environmental Science, 2011, 31(9): 1416-1423. |
| [31] | 胡敏, 何凌燕, 黄晓锋, 等. 北京大气细粒子和超细粒子理化特征、来源及形成机制[M]. 北京: 科学出版社, 2009. |
| [32] | Kulmala M, Petäjä T, Nieminen T, et al. Measurement of the nucleation of atmospheric aerosol particles[J]. Nature Protocols, 2012, 7(9): 1651-1667. DOI:10.1038/nprot.2012.091 |
| [33] | Whitby K T. The physical characteristics of sulfur aerosols[J]. Atmospheric Environment (1967), 1978, 12(1-3): 135-159. DOI:10.1016/0004-6981(78)90196-8 |
| [34] | Zhu Y J, Sabaliauskas K, Liu X H, et al. Comparative analysis of new particle formation events in less and severely polluted urban atmosphere[J]. Atmospheric Environment, 2014, 98: 655-664. DOI:10.1016/j.atmosenv.2014.09.043 |
| [35] | Kulmala M, Petäjä T, Mönkkönen P, et al. On the growth of nucleation mode particles: source rates of condensable vapor in polluted and clean environments[J]. Atmospheric Chemistry and Physics, 2005, 5(2): 409-416. DOI:10.5194/acp-5-409-2005 |
| [36] | Shen X J, Sun J Y, Zhang Y M, et al. First long-term study of particle number size distributions and new particle formation events of regional aerosol in the North China Plain[J]. Atmospheric Chemistry and Physics, 2011, 11(4): 1565-1580. DOI:10.5194/acp-11-1565-2011 |
| [37] | Gao J, Wang J, Cheng S H, et al. Number concentration and size distributions of submicron particles in Jinan urban area: Characteristics in summer and winter[J]. Journal of Environmental Sciences, 2007, 19(12): 1466-1473. DOI:10.1016/S1001-0742(07)60239-3 |
| [38] | Cai R L, Yang D S, Fu Y Y, et al. Aerosol surface area concentration: a governing factor in new particle formation in Beijing[J]. Atmospheric Chemistry and Physics, 2017, 17(20): 12327-12340. DOI:10.5194/acp-17-12327-2017 |
| [39] | Yue D L, Hu M, Wang Z B, et al. Comparison of particle number size distributions and new particle formation between the urban and rural sites in the PRD region, China[J]. Atmospheric Environment, 2013, 76: 181-188. DOI:10.1016/j.atmosenv.2012.11.018 |
| [40] | Dada L, Paasonen P, Nieminen T, et al. Long-term analysis of clear-sky new particle formation events and nonevents in Hyytiälä[J]. Atmospheric Chemistry and Physics, 2017, 17(10): 6227-6241. DOI:10.5194/acp-17-6227-2017 |
| [41] | Hamed A, Korhonen H, Sihto S L, et al. The role of relative humidity in continental new particle formation[J]. Journal of Geophysical Research: Atmospheres, 2011, 116(D3). DOI:10.1029/2010JD014186 |
| [42] | Sogacheva L, Dal Maso M, Kerminen V M, et al. Probability of nucleation events and aerosol particle concentration in different air mass types arriving at Hyytiala southern Finland, based on back trajectories analysis[J]. Boreal Environment Research, 2005, 10(6): 479-491. |
| [43] | Guo H, Wang D W, Cheung K, et al. Observation of aerosol size distribution and new particle formation at a mountain site in subtropical Hong Kong[J]. Atmospheric Chemistry and Physics, 2012, 12(20): 9923-9939. DOI:10.5194/acp-12-9923-2012 |
| [44] | Zhang R Y, Suh I, Zhao J, et al. Atmospheric new particle formation enhanced by organic acids[J]. Science, 2004, 304(5676): 1487-1490. DOI:10.1126/science.1095139 |
| [45] | Yli-Juuti T, Nieminen T, Hirsikko A, et al. Growth rates of nucleation mode particles in Hyytiälä during 2003-2009: variation with particle size, season, data analysis method and ambient conditions[J]. Atmospheric Chemistry and Physics, 2011, 11(24): 12865-12886. DOI:10.5194/acp-11-12865-2011 |
| [46] | Petters M D, Kreidenweis S M. A single parameter representation of hygroscopic growth and cloud condensation nucleus activity[J]. Atmospheric Chemistry and Physics, 2007, 7(8): 1961-1971. DOI:10.5194/acp-7-1961-2007 |
| [47] | Man H Y, Zhu Y J, Ji F, et al. Comparison of daytime and nighttime new particle growth at the HKUST supersite in Hong Kong[J]. Environmental Science & Technology, 2015, 49(12): 7170-7178. |