 2021, Vol. 42
2021, Vol. 42

2. 昆明学院昆明滇池(湖泊)污染防治合作研究中心, 昆明 650214;
3. 昆明市河湖生态健康评估与修复院士工作站, 昆明 650214
 , XU Shan2,3 , ZHAO Zheng2,3 , ZHOU Xiao-hua2,3 , FENG Qing2,3 , YANG Jiang-hua1 , LI Fei-long1 , WANG Zhi-hao1 , ZHANG Xiao-wei1,2,3
, XU Shan2,3 , ZHAO Zheng2,3 , ZHOU Xiao-hua2,3 , FENG Qing2,3 , YANG Jiang-hua1 , LI Fei-long1 , WANG Zhi-hao1 , ZHANG Xiao-wei1,2,3
2. Kunming Dianchi Lake Environmental Protection Collaborative Research Center, Kunming University, Kunming 650214, China;
3. Academician Workstation for Ecological Health Assessment and Rehabilitation of Rivers and Lakes in Kunming, Kunming 650214, China
环境DNA(environmental DNA, eDNA)技术基于环境介质中生物遗传物质的分子序列分析, 可实现生物物种的高效识别与监测, 是本世纪以来生物多样性科学发展中最重要的技术进步. eDNA宏条形码(metabarcoding)技术采用通用引物扩增环境DNA中特定基因片段(即DNA条形码), 借助高通量测序获得DNA条形码序列, 通过与参考物种数据库比对获取水环境中生物的种类及相对丰度[1].与传统形态学监测方法相比, 环境DNA技术具有经济高效, 流程标准化, 重复性好和同时揭示遗传多样性和物种多样性等优势, 是一种潜在补充甚至替代传统形态学的生物监测方法[1, 2].环境DNA宏条形码技术在对生态系统观测与评估的理论研究方面已经展现出极大的作用与价值.然而, 目前尚缺乏对该方法的标准化和规范化研究, 严重限制了其在可考核的生态环境监测体系中的应用与推广.
规范化的eDNA宏条形码监测方法不仅可以实现对生态环境中物种丰富度的精准记录, 而且可实现大尺度生物监测数据的跨时空比较, 助力基于生物指数的水生态健康评价.基于eDNA宏条形码技术生物多样性监测误差受多方面技术参数因素影响, 如采样方法和体积、PCR扩增(引物选择)、测序(测序平台和测序深度)及生物信息学过程(如数据筛选、OTU聚类方法和DNA条形码数据库)等[2].目前不同实验室开展eDNA研究选用的参数均不统一, 难以实现跨实验室监测数据的整合与比较.以真核浮游植物为例, eDNA研究采集的水样体积在50 mL~3 L不等, 但大多数研究的过滤水样小于500 mL, 选择的滤膜孔径范围包括0.22~68 μm[3~6].目前用于真核浮游植物识别的DNA条形码区域包括核基因的ITS、18S rDNA、28S rDNA和叶绿体rbcL基因等[7, 8], 其中18S rDNA基因具有更高的物种覆盖率及较为完善的条形码数据库, 已被广泛应用于监测海洋和淡水真核藻类的组成结构[4, 8, 9].不同研究中样品的测序深度相差超过1个数量级(3 000~100 000条), 不同生态系统的物种丰富度不同, 所需的测序深度也有所不同.目前, SILVA数据库是国际上通用的核糖体小亚基DNA条形码数据库, 其中PR2是专门针对18S rDNA条形码的数据库[10], 也有团队通过构建本土物种DNA条形码数据库以提高eDNA监测物种的准确性[11, 12].因此, 建立统一和规范化的eDNA宏条形码技术监测流程具有重要的意义与价值.
建立eDNA宏条形码生物监测技术的精准度评价方法, 是实现eDNA生物监测数据的质量控制的前提.精准度是衡量监测技术结果可靠性的重要指标, 它不仅体现了数据的稳定性和可信度, 同时会影响跨时间和空间上监测数据的比较, 并影响监测评价结果.精准度包括精确性(重复性)和准确性(真实性), 精确性指重复测量值之间的偏差, 准确性指测量值和真实值之间的差距[13].精确性越高, 证明监测方法更稳定, 监测结果更可信.但是目前我国的水生生物监测研究主要依赖基于形态学的调查方法, 尚未明确系统地提出精确性计算方法及质量控制[13~15].由于真实环境中的生物多样性未知, 因此多用精确性来反映生物多样性监测方法的可靠性.生物监测精确性的检验需同时考虑其生物重复和技术重复.生物重复的精确性即在同一位点采集平行样品, 分别进行实验处理所获得结果的偏差; 技术重复的精确性则指对同一个生物样品做多次相同处理所获得结果的偏差[16].对于单个环境eDNA样品, 宏条形码技术直接获得的数据包括遗传分类单元(OTUs)数及其相对丰度、可注释的物种分类单元及其相对丰度.根据这些技术数据特征, 可计算单个位点物种多样性指数, 包括物种丰富度、香农指数和Pielou均匀度等.这些指数是衡量一个位点生物多样性最为常用的指标, 也被用于评估水生态健康状况, 比如, 通过比较监测位点和参考位点之间的多样性指数, 评价所监测位点水生态的好坏[17~19].因此eDNA监测的精确性评价应包括遗传分类单元、可注释的物种分类单元及相应多样性指数等方面的评估.
本文以高原湖泊滇池和抚仙湖的真核浮游植物为研究对象, 采用eDNA宏条形码生物监测技术监测真核浮游植物多样性的规范化流程, 对两个湖泊的样品进行深度测序, 获取适合滇池和抚仙湖藻类监测的测序深度; 分别在遗传分类单元(OTU)和可注释物种分类单元(属水平)评估eDNA监测结果的精确性; 根据以上研究结果对监测数据进行质量控制, 最终获得滇池和抚仙湖的真核藻类图谱, 对比和形态学监测结果的一致性; 揭示滇池和抚仙湖真核藻类多样性的空间分布差异, 探究并验证eDNA宏条形码技术监测我国湖泊真核浮游植物多样性的可行性.
1 材料与方法 1.1 生物监测位点选择滇池是我国云南省最大的高原浅水型淡水湖泊, 属长江流域金沙江水系.随着周边城市化进程加速, 滇池污染日益严重, 为我国典型的富营养化湖泊[20]; 抚仙湖为云南省第三大高原湖泊, 发源于珠江水系, 为深水型湖泊(最深处≥20 m), 是典型的贫营养性湖泊[21].本研究选取滇池的12个位点及抚仙湖北部的4个位点(FN1~FN4)收集水样(图 1).基于地理和污染特征将滇池的位点分为滇池北(DN1~DN4)、滇池中(DM1~DM4)和滇池南(DS1~DS4)这3个区域.其中滇池北部以建成的昆明主城区为主, 滇池中部主要为正在建设的呈贡新区及旅游业为主, 滇池北部则主要以农业种植为主[22].

|
图 1 采样位点分布示意 Fig. 1 Sampling sites in the study area |
具体的研究过程参照图 2.

|
图 2 本研究技术路线示意 Fig. 2 Flow chart for this study |
每个点位采集水样1 L.为了分析不同水层的物种组成差异, 滇池DN1位点分别于水下0.5 m(表层)、2 m(中层)和4 m(底层)采集水样; 抚仙湖FN1位点于水下0.5 m(表层)、10 m(中层)和20 m(底层)分别采集水样.每升水样分别过滤到3张0.45 μm的亲水性尼龙膜(Merck Millipore, 美国), 即每张滤膜过滤水样330 mL, 每个点位3个生物重复.富集环境DNA的滤膜分别装入5 mL的离心管中, -20℃保存.
1.2.2 DNA提取及PCR扩增采用DNeasy PowerWater Kit试剂盒(QIAGEN, 德国)提取环境DNA.利用通用引物1380F和1510R扩增真核浮游植物18S rDNA基因的V9区域, 扩增引物为1380F: 5′-TCCCTGCCHTTTGTACA CAC-3′, 1510R: 5′-CCTTCYGCAGGTTCACCTAC-3′[23]. PCR扩增体系为50 μL: 25 μL Phanta聚合酶(诺唯赞), 10 μmol·L-1上游和下游引物各1 μL, 2 μL的模板DNA和21 μL的无酶水. PCR反应在SureCycler 8800热循环仪(Agilent Technologies, USA)上进行.反应程序为98℃预变性30 s, 25个循环反应, 98℃变性10 s, 59℃退火20 s, 72℃延伸10 s. PCR产物经1.5%琼脂糖凝胶电泳检测条带, 之后用E.Z.N.A.TM E-Z 96×5 Cycle Pure Kit (OMEGA)试剂盒纯化样品, 用Qubit 2.0测定纯化后DNA浓度.
1.2.3 文库构建及测序每个纯化后PCR产物根据100 ng等量混合, 取混库后的DNA样品再次纯化Agencourt AMPure XP kit (Becman Coulter, 美国), 利用Ion Plus Fragment Library Kit试剂盒(Life Technologies, 美国)构建测序文库. 2100 Bioanalyzer (Agilent Technologies, 美国)检测DNA文库质量.将测序文库稀释至100 pmol·L-1, 采用试剂盒Ion 540TM Kit-OT2(Life Technologies, 美国)将测序文库连接到ISPs(ion sphere particles)的表面, 用Ion S5TM测序仪(Life Technologies, 美国)测序, 保证每个样品的序列数不少于4万条.
1.2.4 生物信息学分析利用软件MOTHUR将FASTQ格式的文件转为FASTA和对应的质量文件; 在综合计算平台QIIME(quantitative insights into microbial ecology v1.8.0)上根据barcode不同分割样品序列, 进一步去除测序错误率高于1%, 长度小于100 bp及出现次数小于2的序列.用USEARCH7按照97%的相似性将序列聚类成可操作分类单元(operational taxonomic units, OTUs), 并将其比对到公共数据库去除嵌合体, 序列比对归类到每个OTU, 获得每个OTU在每个样品中的序列数.基于Python语言对OTUs根据PR2数据库(https://github.com/pr2database/pr2database)进行物种注释[24], 注释到真核藻类各个门的OTUs定义为真核藻类OTUs, 进一步根据文献[25]中的藻类物种名录, 筛选中国淡水真核藻类OTUs.
1.3 统计分析 1.3.1 α多样性指数分析在R语言中Bioconductor环境下, 用软件包“vegan”计算真核藻类群落的丰富度S、
香农指数[Shannon指数, 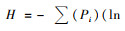
分别选择滇池DN1和抚仙湖FN1位点的表层水样进行深度测序, 每个样品序列数均大于8万条.在R语言中用“vegan”包对每个样品序列按照2 000的等差测序深度随机抽样100次, 获得每个测序深度对应的真核藻类OTU或属数目, 在Graphpad Prism 8中绘图.
1.3.3 精确性评价指标计算(1) 平行间OTU或属的交叉情况

|
(2) α多样性的变异系数(CV值)
分别基于生物重复在OTU水平和属水平的丰富度, 香农指数和Pielou均匀度计算变异系数.
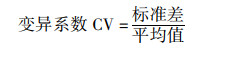
|
单因素方差分析不同水深及不同区域生物多样性指数的差异, 在Graphpad Prism 8中绘图.
2 结果与分析测序共获4 426 086条原始序列, 对原始序列过滤处理, 获得高质量序列4 392 050条. DN1和FN1表层水6个样品的平均序列89 560条, 其余样品序列分布在41 642~47 000条.对高质量序列进行OTU聚类, 共获得3 960个OTUs, 其中有2 658个OTUs能被数据库注释, 属于真核浮游藻类的OTU共1 040个, 占总注释OTUs的39.13%.有765个OTU能被注释到属水平, 占总序列数的32.40%.
2.1 测序深度对OTU和属分类单元数目的影响对典型位点eDNA样品宏条形码深度测序结果的分析表明, 随着测序深度的增加, 获得的OTU和属数目均在不断增加, 最后趋于平缓.滇池D1监测到266个真核藻类OTU, 36 000和40 000的测序深度可分别捕捉90%和95%的OTU, 当测序深度达到38 000, 稀释性曲线基本饱和.抚仙湖FN1点位共发现OTU数目374个, 当测序深度达到30 000和36 000, 检出OTU的数目分别达到90%和95%, 当测序深度达到36 000, 稀释性曲线基本饱和.滇池DN1点位平均识别真核藻类62个属, 90%和95%以上的属分别需要的测序深度是30 000和38 000条序, 抚仙湖FN1位点的OTU平均注释到83个真核藻属, 测序深度为20 000和28 000可分别获得90%和95%的属(图 3).

|
图 3 真核藻类分类单元数目随着测序深度的变化 Fig. 3 Taxon number of Eukaryotic phytoplankton at different sequencing depths |
3个生物平行样品中均出现的OTU交叉率达到45.97%±1.67%, 至少在两个平行样中出现的OTU所占比例为68.01%±1.98%. 3个平行样品考虑序列数目权重的OTU交叉率达到92.83%±2.74%(表 1).
|
|
表 1 eDNA宏条形码监测数据的精确性评价结果/% Table 1 Biodiversity Precision assessment based on eDNA metabarcoding data/% |
单因素方差分析显示生物重复之间的OTU多样性无显著差异(P>0.1).基于多样性指数计算生物重复之间OTU多样性的CV值, 丰富度的CV值为7.54%±2.60%, 香农指数和Pielou均匀度的CV值分别为2.82%±1.70%和3.55%±1.59%, 均低于5% [表 1和图 4(a)].

|
图 4 基于OTU和属的α多样性指数的精确性 Fig. 4 Degree of precision of α biodiversity index |
在属水平上, 3个平行样品的交叉率达到64.21%±3.25%, 至少出现在两个平行样的属所占比例为84.16%±3.76%(表 1).若考虑序列数目权重, 则3个平行样品的属交叉率达到98.41%±0.51%.
单因素方差分析显示生物重复之间的属α多样性无显著性差异(P>0.1).在属水平, 丰富度的CV值为4.71%±2.30%, 香农指数和Pielou均匀度的CV值分别为2.51%±0.98%和3.63%±0.95%, 均小于5% [表 1和图 4(b)].
2.2.3 测序深度影响OTU或属交叉率Pearson相关性分析显示在测序深度小于30 000时, 序列数和分类单元的交叉率显著正相关, 其中与OTU水平的相关性更加明显(OTU水平:R2=0.527, P < 0.000 1;属水平:R2=0.288, P < 0.000 3).随着测序深度的增加, 交叉率也会逐渐平稳饱和.当序列数为2 000时, 平行间OTU交叉率为26.81%±4.37%, 当序列数达到30 000时, OTU交叉率为42.69%±1.78%, 基本达到饱和.属交叉率明显高于OTU交叉率, 当测序深度为2 000时, 平行间属交叉率为41.46%±6.56%, 测序深度达到22 000时, 属交叉率达超过67%, 变化趋于平缓(图 5).

|
图 5 生物重复间OTU或属水平的交叉率随测序深度的变化 Fig. 5 Overlap of OTU or genera among biological replicates at different sequencing depths |
eDNA宏条形码共检出真核藻类110属, 分属于9门16纲32目66科, 其中绿藻门的种类最丰富, 共识别出30科53属, 在种类数量上占有绝对优势, 和已有形态学研究结果一致.角甲藻、衣藻和多甲角藻识别出的OTUs数目最多, 冠盘藻和小环藻属次之.冠盘藻、栅藻和裸甲藻的序列数最多, 占真核藻类序列数的30%以上, 其次为衣藻和隐藻属(图 6).

|
(a)基于条形码序列的系统进化关系, 进化距离依据K2P模型计算, 进化树构建基于邻接法; (b)每个属对应的OTU数目; (c)每个属对应的序列数; (d)属的检出频率; (e)属在每个点位的分布情况, 点的大小代表特定属在该点位的相对丰度 图 6 eDNA宏条形码监测表征的真核浮游植物群落 Fig. 6 Eukaryotic phytoplankton community composition characterized using eDNA metabarcoding |
滇池共监测到真核藻类75个属, 分属于8门13纲23目49科.其中绿藻门和硅藻门为主要优势类群, 绿藻门有33属, 占总序列数的40.27%, 共识别硅藻14属, 占总序列数的41.67%.优势属为冠盘藻, 栅藻和隐藻属(图 6).
抚仙湖10个位点监测到真核藻类90个属, 分属于9门13纲29目56科, 绿藻门和硅藻门物种丰富度最高, 绿藻门包含42个属, 硅藻门16个属.甲藻门相对丰度最高, 占总序列的40.94%, 其次为硅藻门.绝对优势种为裸甲藻属和衣藻属(相对丰度>10%), 其次为栅藻, 多甲藻, 原甲藻和金色藻属(图 6).
2.3.2 eDNA宏条形码和传统形态学监测结果的比较eDNA宏条形码覆盖了滇池62.5%和抚仙湖71.05%的形态学监测数据, 表明两种方法监测结果具有较高一致性(图 7).同时eDNA宏条形码检测到金藻门, 褐藻门和定鞭藻门等显微镜鉴定未识别的类群. eDNA识别滇池的真核藻类75属, 包含王华等[20]于2016年在滇池相似位点识别出24属真核藻类的15属.抚仙湖eDNA识别90个真核藻类的属, 覆盖了形态学识别出的38个真核藻属的27种[26].滇池和抚仙湖通过传统形态学监测到的共有属19种, 其中14种能被eDNA识别.

|
DC:滇池FXH:抚仙湖; T:形态学监测结果; eDNA:环境DNA宏条形码监测结果; 括号外、内数字分别表示该区域的属数目及所占比例 图 7 eDNA宏条形码和形态学监测真核浮游植物在属水平的比较 Fig. 7 Comparison of metabarcoding data with traditional monitoring data at the genus level |
滇池DN1位点不同水深的真核藻类多样性无明显差异, 抚仙湖FN1位点的真核藻类多样性具有显著垂直分布特征.滇池属于浅水型湖泊, 分别取0.5、2和4 m水深的样品进行生物多样性分析, 基于OTU计算的真核藻类香农指数存在显著性差异(P=0.015), 其余多样性参数无明显差异(P>0.05).基于抚仙湖FN1点位的不同水深OTU数据获得的丰富度, 香农指数和Pielou均匀度均存在显著性差异(P < 0.05), 10 m左右水层真核藻类的种类最丰富, 生物多样性最高, 均匀度也略高, 其次为表层水.在可注释分类单元属水平, 中层水(10 m)的真核藻类多样性显著高于表层水和底层水, 但是level表层水和底层水的生物多样性无显著差异(图 8).

|
(a)~(c)基于OTU的生物多样性差异; (d)~(e)基于可注释分类单元属的生物多样性差异; ns:无显著性差异(P>0.05);不同字母表示显著性差异(P < 0.05) 图 8 滇池和抚仙湖不同水深的生物多样性差异 Fig. 8 Differences in biodiversity determined at different depths at the OTU and genus level |
抚仙湖北的生物多样性明显高于滇池各区域, 滇池南部的生物多样性明显高于中部和北部.基于OTU和属分类单元的真核藻类多样性结果均显示, 滇池南部的丰富度和香农指数明显高于滇池中部和北部, 滇池北和滇池中部的差异不显著.抚仙湖北的真核藻类的OTU和属对应的香农指数分别为4.55±0.73和2.92±0.17, 生物多样性明显高于滇池不同区域(P < 0.05, 图 9).

|
(a)~(c)基于OTU的生物多样性差异; (d)~(f)基于可注释分类单元属的生物多样性差异; **代表P < 0.01;不同字母表示显著性差异(P < 0.05) 图 9 不同区域的生物多样性差异 Fig. 9 Biodiversity differences among regions |
深度测序通常会提高物种多样性的监测力, 但是随着测序深度的增加, 成本也会不断增加, 因此选择合适的测序深度, 是eDNA监测方法需要进行质量控制的关键指标.滇池和抚仙湖的真核藻属的稀释性曲线达到饱和所需要的测序深度分别为38 000和36 000.考虑到测序成本, 可选择获得90%属对应的测序深度, 即滇池和抚仙湖位点分别需要30 000和20 000条序列.另外, 测序深度增加, OTU及属分类单元交叉率也会提高, 监测数据的精确性提高, 因此同时考虑属分类单元数目及交叉率, 则滇池北和抚仙湖北需要的测序深度分别为30 000和22 000条序列.滇池真核藻类监测所需的测序深度高于抚仙湖, 这说明不同生态系统的生物多样性的复杂程度不同, 所需的测序深度也略有差别.同时两个湖泊的真核藻属的稀释性曲线达到饱和所需要的测序深度, 高于已有其他类群(如浮游动物)的eDNA监测所需要的测序通量[27], 这可能和生物类群多样性的固有差异相关, 也可能是由于18S-V9引物扩增的序列中包含大量非藻类的序列, 导致序列利用率下降.
3.2 生物多样性监测的精确性评价为了确保监测结果的可靠性, 我国针对不同的监测指标, 分别制定了质控措施, 对监测方法和仪器的精密度与准确度提出明确要求.环境DNA宏条形码技术作为一种新型生物多样性监测方法, 已广泛应用于水生态系统中的不同群落多样性研究, 但要将其推广应用到监测和管理部门, 需要充分保证监测数据的准确性.本研究通过平行样分析eDNA监测结果的可重复性, 分别从遗传多样性OTU和可注释分类单元属水平构建eDNA监测精确性评价方法, 评价指标包括重复之间OTU或属的交叉率及α多样性指数的变异系数.
物种分类单元的交叉率是目前评价eDNA监测结果重复性最为常用的方法.已有基于454焦磷酸测序平台的研究发现重复样本间OTU交叉率为59.8%, 在目水平的交叉率可达到89.5%, 说明更高的物种分类单元交叉率更高[28].本研究中生物重复之间OTU交叉率为45.97%±1.67%, 属交叉率达到64.21%±3.25%, 考虑序列数目权重的OTU和属交叉率分别达到92.83%±2.74%和98.41%±0.51%. Wen等[17]基于Illumina测序平台, 用16S-V4引物扩增土壤中微生物多样性, 发现不同测序重复之间在OTU水平的重复率为39%, 但是基于序列数目权重的OTU交叉率达到99%, 和本研究结果基本一致, 说明平行样品之间交叉的OTU覆盖了高丰度的OTU, 每个平行样品特有的OTU为低丰度OTU(如只具有1或2个序列的OTUs).有研究认为这些低丰度OTUs可能是实验室操作过程中引入的测序错误, 如PCR扩增和测序过程中引入的低频率错误序列[17], 但是也有研究发现这些低丰度的OTUs可能是群落中的稀有物种, 具有重要的生态功能[29].因此, 数据分析过程中, 应该慎重选择OTU的筛选阈值.
物种丰富度, 香农指数和Pielou均匀度是评估一个点位生物多样性和水生态健康状况最为常用的指标, 本研究首先通过单因素方差分析, 发现生物重复之间的误差, 不会显著影响α多样性.进一步比较了生物重复之间的变异系数, 发现物种丰富的CV值< 10%, 香农指数和Pielou均匀度的CV值< 5%, 进一步说明eDNA监测结果的可靠性和稳定性.
平行样品间的生物多样性监测误差受多方面因素影响:①采样不均匀; ②技术误差, 如DNA提取(DNA裂解效率), PCR扩增(引物扩增偏好性及扩增条件), 测序(测序平台的选择, 测序深度及固有误差)及生物信息学过程(如数据筛选, OTU聚类方法选择, 嵌合子过滤等); ③采样或实验室污染引入少量低丰度的序列误差等[16, 17].因此在eDNA监测过程中需要采集平行样品, 同时针对每个处理步骤做好质控, 也可针对每个过程设置对照, 来识别和降低误差, 保证数据质量和评估结果的可信度.
3.3 eDNA监测真核浮游植物多样性的可行性 3.3.1 eDNA宏条形码和形态学监测结果的一致性滇池和抚仙湖北各监测到真核藻类75属和90属, 分别覆盖了62.5%和71.05%形态学监测到的真核藻类[20, 26], 其中eDNA识别的滇池(如栅藻属、隐藻属和冠盘藻属等)和抚仙湖(如衣藻属, 锥囊藻属和脆杆藻属等)的优势属, 在以往的形态学调查中也作为优势类群出现[20, 30, 31], 说明eDNA监测较高的还原力.原理上, 任意两个物种都存在基因序列的差异, 因此形态学可监测到的物种几乎都可以被DNA宏条形码方法识别出来[7].目前DNA宏条形码未监测出来的物种, 有很大一部分缺乏相应的DNA条形码数据, 这一问题将会随着参考数据库的不断补充而解决[32].相比于传统形态学, eDNA宏条形码可以识别出更多的物种, 尤其对于稀有物种和隐蔽物种, 如金藻门和褐藻门.已有大量研究也发现很多隐匿种及形态学分类特征不明显的物种可以被宏条形码技术检测到[2, 8, 33], 说明基于分子水平的物种识别方面具有更高的灵敏度和分辨率.
3.3.2 eDNA宏条形码技术可以表征不同空间尺度真核藻类多样性的差异垂直空间调查显示, 滇池不同水深真核藻类多样性差别不大, 但是抚仙湖不同水深的真核藻类多样性具有显著性差异, 且抚仙湖10 m水深的生物多样性明显高于表层水和深层水,
这和已有基于形态学的研究结果一致[34].这是由于抚仙湖是深水型湖泊, 夏季具有明显的温跃层, 抑制了水体的上下混合交换, 进而导致浮游植物的垂直分布差异[35].水平空间调查显示, 抚仙湖北真核浮游植物的生物多样性高于富营养化的滇池, 这可能是由于抚仙湖生态系统的生产力更低, 营养资源有限, 物种之间竞争更弱, 从而保证更多的物种共存, 生物多样性增加[36].滇池南部的真核藻类多样性明显高于滇池北部和中部, 可能由于人类活动导致不同区域的污染类型程度存在差异, 影响了浮游植物的多样性.
4 结论(1) 本研究通过分析平行样本间遗传分类单元和属分类水平的交叉率及α多样性的精确性两个指标, 评估了eDNA监测真核浮游植物多样性的准确性.当真核藻类α多样性的CV值< 10%, 且至少在两个重复之间出现的OTU或属的交叉率>50%时, 说明平行生物样品间具有良好的重复性, 可将同一位点不同重复之间的结果合并, 用于后续点位甚至区域之间生物多样性比较.
(2) 测序深度显著影响监测到的物种分类单元数目及精确性评估结果, 不同水体由于物种组成差异, 所需要的测序通量存在差异.本研究中, eDNA宏条形码18S-V9监测高原湖泊滇池和抚仙湖的真核藻属, 需要的测序深度不少于30 000条序列.
(3) eDNA宏条形码技术显著提高了两个典型湖泊中真核浮游植物的物种监测效率. eDNA监测到的真核藻属覆盖了传统形态学监测的62%以上, 同时eDNA可以检测到更多传统形态学尚未识别的真核藻属, 具有更高的灵敏性.另外eDNA宏条形码监测结果显示, 在滇池和抚仙湖北不同区域间存在显著的生物多样性差异, 表明该技术可以表征和区分不同空间尺度的真核浮游植物多样性, 具有高分辨率.
(4) 本研究证实新型的eDNA宏条形码技术可应用于湖泊真核浮游植物多样性监测.和传统形态学相比, eDNA宏条形码监测具有多方面优势:如采样可在时间上提高采样频率, 空间上设置更多的监测位点, 可用于真核浮游植物群落的跨生态系统和跨时间比较.为进一步推动eDNA宏条形码监测技术的规范化, 未来可开展跨实验室比对验证实验, 通过采用统一的eDNA宏条形码标准化分析流程, 在不同实验室由不同操作者进行重复, 对监测结果进行验证比较; 并在推广实践过程中需要评估监测结果的准确性, 并且做好质量控制.
| [1] | Zhang X W. Environmental DNA Shaping a new era of ecotoxicological research[J]. Environmental Science & Technology, 2019, 53(10): 5605-5612. |
| [2] | Debroas D, Domaizon I, Humbert J F, et al. Overview of freshwater microbial eukaryotes diversity: a first analysis of publicly available metabarcoding data[J]. FEMS Microbiology Ecology, 2017, 93(4). DOI:10.1093/femsec/fix023 |
| [3] | Machado K B, Targueta C P, Antunes A M, et al. Diversity patterns of planktonic microeukaryote communities in tropical floodplain lakes based on 18S rDNA gene sequences[J]. Journal of Plankton Research, 2019, 41(3): 241-256. DOI:10.1093/plankt/fbz019 |
| [4] | Reboul G, Moreira D, Bertolino P, et al. Microbial eukaryotes in the suboxic chemosynthetic ecosystem of Movile Cave, Romania[J]. Environmental Microbiology Reports, 2019, 11(3): 464-473. DOI:10.1111/1758-2229.12756 |
| [5] | Tragin M, Vaulot D. Novel diversity within marine Mamiellophyceae (Chlorophyta) unveiled by metabarcoding[J]. Scientific Reports, 2019, 9(1). DOI:10.1038/s41598-019-41680-6 |
| [6] | Sze Y, Miranda L N, Sin T M, et al. Characterising planktonic dinoflagellate diversity in Singapore using DNA metabarcoding[J]. Metabarcoding and Metagenomics, 2018, 2(1). DOI:10.3897/mbmg.2.25136 |
| [7] |
张宛宛, 谢玉为, 杨江华, 等. DNA宏条形码(metabarcoding)技术在浮游植物群落监测研究中的应用[J]. 生态毒理学报, 2017, 12(1): 15-24. Zhang W W, Xie Y W, Yang J H, et al. Applications and prospects of metabarcoding in environmental monitoring of phytoplankton community[J]. Asian Journal of Ecotoxicology, 2017, 12(1): 15-24. |
| [8] |
王靖淇, 王书平, 张远, 等. 高通量测序技术研究辽河真核浮游藻类的群落结构特征[J]. 环境科学, 2017, 38(4): 1403-1413. Wang J Q, Wang S P, Zhang Y, et al. Community structure characteristics of eukaryotic planktonic algae in Liaohe River through high-throughput sequencing[J]. Environmental Science, 2017, 38(4): 1403-1413. |
| [9] | 张宛宛.基于DNA宏条形码技术的浮游植物群落多样性监测研究[D].南京: 南京大学, 2017. |
| [10] | Guillou L, Bachar D, Audic S, et al. The protist ribosomal reference database (PR2): a catalog of unicellular eukaryote small sub-unit rRNA sequences with curated taxonomy[J]. Nucleic Acids Research, 2013, 41(D1): D597-D604. |
| [11] | Decelle J, Romac S, Stern R F, et al. PhytoREF: a reference database of the plastidial 16S rRNA gene of photosynthetic eukaryotes with curated taxonomy[J]. Molecular Ecology Resources, 2015, 15(6): 1435-1445. DOI:10.1111/1755-0998.12401 |
| [12] | Tragin M, Vaulot D. Green microalgae in marine coastal waters: the Ocean Sampling Day (OSD) dataset[J]. Scientific Reports, 2018, 8. DOI:10.1038/s41598-018-32338-w |
| [13] |
Markert B, 王美娥, Wünschmann S, 等. 环境质量评价中的生物指示与生物监测[J]. 生态学报, 2013, 33(1): 33-44. Bernd M, Wang M E, Wünschmann S, et al. Bioindicators and biomonitors in environmental quality assessment[J]. Acta Ecologica Sinica, 2013, 33(1): 33-44. |
| [14] |
阴琨, 王业耀, 许人骥, 等. 中国流域水环境生物监测体系构成和发展[J]. 中国环境监测, 2014, 30(5): 114-120. Yin K, Wang Y Y, Xu R J, et al. Constitution and development of national river basin water environmental biomonitoring system[J]. Environmental Monitoring in China, 2014, 30(5): 114-120. DOI:10.3969/j.issn.1002-6002.2014.05.025 |
| [15] |
李黎, 王瑜, 林岿璇, 等. 河流生态系统指示生物与生物监测:概念、方法及发展趋势[J]. 中国环境监测, 2018, 34(6): 26-36. Li L, Wang Y, Lin K X, et al. Bioindicator and biomonitoring used for river ecosystem: Concepts, approaches and trends[J]. Environmental Monitoring in China, 2018, 34(6): 26-36. |
| [16] | Zhan A B, He S, Brown E A, et al. Reproducibility of pyrosequencing data for biodiversity assessment in complex communities[J]. Methods in Ecology and Evolution, 2014, 5(9): 881-890. DOI:10.1111/2041-210X.12230 |
| [17] | Wen C Q, Wu L Y, Qin Y J, et al. Evaluation of the reproducibility of amplicon sequencing with Illumina MiSeq platform[J]. PLoS One, 2017, 12(4). DOI:10.1371/journal.pone.0176716 |
| [18] |
秦娇娇, 王艳. 浮游植物多样性指数的应用及评价[J]. 沈阳师范大学学报(自然科学版), 2014, 32(4): 502-505. Qin J J, Wang Y. Application and evaluation of phytoplankton diversity indexes[J]. Journal of Shenyang Normal University (Natural Science Edition), 2014, 32(4): 502-505. DOI:10.3969/j.issn.1673-5862.2014.04.010 |
| [19] |
顾礼明, 曹慧敏, 范铭芳, 等. 大型湖库浮游植物生物多样性研究进展综述[J]. 污染防治技术, 2019, 32(4): 37-39. Gu M L, Cao H M, Fan M F, et al. Research progress of phytoplankton biodiversity in large lakes and reservoirs: a review[J]. Pollution Control Technology, 2019, 32(4): 37-39. |
| [20] |
王华, 杨树平, 房晟忠, 等. 滇池浮游植物群落特征及与环境因子的典范对应分析[J]. 中国环境科学, 2016, 36(2): 544-552. Wang H, Yang S P, Fang S Z, et al. Canonical correspondence analysis of relationship between characteristics of phytoplankton community and environmental factors in Dianchi Lake[J]. China Environmental Science, 2016, 36(2): 544-552. DOI:10.3969/j.issn.1000-6923.2016.02.034 |
| [21] |
董云仙. 云南九大高原湖泊藻类研究进展[J]. 环境科学导刊, 2014, 33(2): 1-8. Dong Y X. Research and development of algae in the nine plateau lakes in Yunnan[J]. Environmental Science Survey, 2014, 33(2): 1-8. DOI:10.3969/j.issn.1673-9655.2014.02.001 |
| [22] |
凌祯, 史正涛, 徐杉, 等. 滇池湖滨带叶绿素a与营养盐空间分布特征[J]. 水文, 2020, 40(1): 76-80. Ling Z, Shi Z T, Xu S, et al. Spatial distribution characteristics of chla and nutrient Salt in Dianchi lakeside[J]. Journal of China Hydrology, 2020, 40(1): 76-80. |
| [23] | Amaral-Zettler L A, McCliment E A, Ducklow H W, et al. A method for studying protistan diversity using massively parallel sequencing of V9 hypervariable regions of small-subunit ribosomal RNA genes[J]. PLoS One, 2009, 4(12). DOI:10.1371/annotation/50c43133-0df5-4b8b-8975-8cc37d4f2f26 |
| [24] | Morard R, Darling K F, Mahé F, et al. PFR2: a curated database of planktonic foraminifera 18S ribosomal DNA as a resource for studies of plankton ecology, biogeography and evolution[J]. Molecular Ecology Resources, 2015, 15(6): 1472-1485. DOI:10.1111/1755-0998.12410 |
| [25] |
胡鸿钧, 魏印心. 中国淡水藻类——系统、分类及生态[M]. 北京: 科学出版社, 2006. Hu H J, Wei Y X. The freshwater algae of China-systematics, taxonomy and ecology[M]. Beijing: Science Press, 2006. |
| [26] | 孔祥虹.抚仙湖浮游植物的时空格局[D].武汉: 湖北大学, 2015. |
| [27] | Yang J H, Zhang X W, Xie Y W, et al. Zooplankton community profiling in a eutrophic freshwater ecosystem-Lake Tai basin by DNA metabarcoding[J]. Scientific Reports, 2017, 7(1). DOI:10.1038/s41598-017-01808-y |
| [28] | Zhang A B, He S, Brown E A, et al. Reproducibility of pyrosequencing data for biodiversity assessment in complex communities[J]. Methods in Ecology and Evolution, 2014, 5(9): 881-890. DOI:10.1111/2041-210X.12230 |
| [29] | Jousset A, Bienhold C, Chatzinotas A, et al. Where less may be more: how the rare biosphere pulls ecosystems strings[J]. The ISME Journal, 2017, 11(4): 853-862. DOI:10.1038/ismej.2016.174 |
| [30] |
吉正元, 刘绍俊, 施艳峰, 等. 抚仙湖浮游植物群落结构周年变化及水质生物评价[J]. 海洋湖沼通报, 2019(3): 78-86. Ji Z Y, Liu S J, Shi Y F, et al. Annual variation of phytoplankton community structure in Fuxian Lake and bioassessment of its water quality[J]. Transactions of Oceanology and Limnology, 2019(3): 78-86. |
| [31] |
张梅, 李原, 王若南. 滇池浮游植物种类的动态变化[J]. 云南大学学报(自然科学版), 2006, 28(1): 73-77. Zhang M, Li Y, Wang R N. Dynamic variation for the species of phytoplankton in Dianchi Lake, China[J]. Journal of Yunnan University (Natural Sciences Edition), 2006, 28(1): 73-77. |
| [32] | Abad D, Albaina A, Aguirre M, et al. Is metabarcoding suitable for estuarine plankton monitoring? A comparative study with microscopy[J]. Marine Biology, 2016, 163(7). DOI:10.1007/s00227-016-2920-0 |
| [33] | Leblanc K, Quéguiner B, Diaz F, et al. Nanoplanktonic diatoms are globally overlooked but play a role in spring blooms and carbon export[J]. Nature Communications, 2018, 9(1). DOI:10.1038/s41467-018-03376-9 |
| [34] |
牛远, 孔祥虹, 余辉, 等. 抚仙湖夏季热分层时期浮游植物空间分布特征[J]. 生态学杂志, 2016, 35(7): 1865-1871. Niu Y, Kong X H, Yu H, et al. Spatial distribution of phytoplankton community during summer stratification in Lake Fuxian[J]. Chinese Journal of Ecology, 2016, 35(7): 1865-1871. |
| [35] |
潘继征, 熊飞, 李文朝, 等. 抚仙湖浮游植物群落结构、分布及其影响因子[J]. 生态学报, 2009, 29(10): 5376-5385. Pan J Z, Xiong F, Li W C, et al. Structure, distribution and its impact factors of phytoplankton community in Fuxian Lake[J]. Acta Ecologica Sinica, 2009, 29(10): 5376-5385. DOI:10.3321/j.issn:1000-0933.2009.10.024 |
| [36] |
陈小林, 陈光杰, 卢慧斌, 等. 抚仙湖和滇池硅藻生物多样性与生产力关系的时间格局[J]. 生物多样性, 2015, 23(1): 89-100. Chen X L, Chen G J, Lu H B, et al. Long-term diatom biodiversity responses to productivity in lakes of Fuxian and Dianchi[J]. Biodiversity Science, 2015, 23(1): 89-100. |