 2021, Vol. 42
2021, Vol. 42



湖泊富营养化已经成为全球性的环境问题[1, 2].磷(P)作为引起水体富营养化的限制性因素, 控制磷污染物进入水体便成为了水体富营养化控制的关键[3, 4].水体磷污染物的来源可分为外源输入和内源输入[5].近年来, 随着政府对环境保护的重视以及民众环保意识的不断加强, 外源磷污染已经被有效地遏制, 但水体的富营养化状况并未得到有效地解决[6], 这主要是因为沉积物与上覆水之间的磷循环(内源磷负荷)仍会不断释放内源磷长达数十年[7, 8].因此, 近年的研究提出许多方法来减少内源磷负荷, 包括底泥疏浚[9]、底泥曝气[10]和原位固定[11]等.
原位固定技术是控制富营养化湖泊内源磷释放的有效方法[12], 其原理是向沉积物中添加磷固定剂, 通过化学吸附、絮凝、沉淀或化学物质与沉积物中各种形态磷发生化学反应而稳定固化, 将沉积物中的潜在活性磷(PMP)转化为更稳定的磷[13, 14].然而, 有研究发现, 原位固定技术虽然对内源磷具有良好的控制效果, 但效果仅限于沉积物表层, 难以影响深层沉积物[15]; 另外, 磷固定剂会阻止溶解氧渗透到沉积物中, 加剧沉积物厌氧环境, 从而导致深层沉积物中磷的积累.因此, 利用原位固磷技术对深层内源磷进行有效控制是当前原位修复研究的重点和热点[16].
本研究通过原位模拟实验, 以氢氧化钙[Ca(OH)2]为磷固定剂, 过氧化氢(H2 O2)为氧化剂, 以梅花散点注射的方式, 考察氧化剂对磷固定剂原位固磷效果的影响.本研究采用薄膜扩散梯度(DGT)和微电极高分辨率采样技术, 分析了不同处理下沉积物中溶解性磷酸盐的细微变化; 通过比较不同处理下沉积物中氮、磷的影响变化, 考察氧化剂存在下固磷材料的纵向影响变化; 探讨不同处理方法对沉积物形态磷变化影响, 以期对原位固磷技术的发展和改良提供数据支持和理论参考.
1 材料与方法 1.1 实验材料本实验所用的氢氧化钙[Ca(OH)2]、双氧水(30%)、浓硫酸、浓盐酸、钼酸氨和抗坏血酸等药剂均为分析纯, 购自中国国药化学药剂有限公司. Rhizon孔隙水采样器和薄膜扩散DGT均由Easysensor Ltd.(中国南京)提供. DGT在使用前用氮气去氧16 h.
1.2 研究地点及采样原位模拟沉积物利用抓斗式沉积物采样器(张口面积约625 cm2)在苏州市某典型富营养化河道采样点(N31°31′30.10″, E120°56′29.7″)采集表层约8 cm沉积物, 同时采集上覆水50 L.采样结束迅速运回实验室, 运输过程中冷藏, 避光, 尽量不扰动.将收集到的沉积物进行混合均匀化, 然后通过0.6 mm孔径的网筛去除大颗粒和大型动物群, 4℃阴暗保存.取一定量的沉淀物, 经冻干、压碎, 再通过100目筛网, 4℃阴暗保存, 直到进一步分析.
1.3 实验方法沉积物样品置于有机玻璃管中(直径84 cm, 高度20 cm), 形成一个15 cm厚度的沉积物层.水样经0.45 μm硝酸纤维素滤膜(Millipore)过滤后, 虹吸入原位装置内, 避免因沉积物悬浮而造成的误差.水层厚度约为10 cm.每个原位装置均插入6根Rhizon孔隙水采样器收集孔隙水.采样器安装在沉积物-水界面以下深度为0(沉积物-水界面)、10、20、30、40、50和60 mm的预成型孔中.
将沉积物岩心分为3组, 每组按不添加任何药剂的对照(Control组)、单一注射氢氧化钙[Ca(OH)2组]和氢氧化钙与双氧水混合注射[OX+Ca(OH)2组]. Ca2+的注射剂量为50 Ca2+/Pmobile(Ca2+用量与表层60 mm沉积物中流动P的摩尔比); H2 O2的施用剂量为50 H2 O2/Pmobile(H2 O2用量与表层60 mm沉积物中流动P的摩尔比).药剂均与过滤后的上覆水混合后, 缓慢注入沉积物深度20 mm处.每隔数小时采集沉积物孔隙水(添加处理剂后一次, 0 h), 每个时间点用Rhizon孔隙水采样器收集孔隙水约1 mL, 采集后立即测定SRP.每次取水样结束后, 立即以虹吸方式补入等量原上覆水.Zr-oxide DGT被用于二维测量沉积物-水剖面中的不稳定磷.所有DGT在原位模拟结束后垂直插入到沉积物中, 24 h后取出. DGT表面用去离子水冲洗, 然后放入干净的自密封袋中等待分析.本实验结束后将沉积物表层切分为3层(Ⅰ层0~20 mm、Ⅱ层20~40 mm和Ⅲ层40~60 mm), 分析各层的形态磷含量.沉积物剖面ORP采用微电极系统(Unisense, 丹麦), 用尖端细至100 μm的克拉克电极测定.
1.4 分析方法上覆水和间隙水中的SRP和NH4+-N分别采用钼锑抗分光光度法和纳氏试剂分光光度法测定.沉积物形态磷分为易交换态磷(或弱吸附态磷)(NH4Cl-P)、铁结合态磷(BD-P)、铝结合态磷(Al-P)、有机磷(Org-P)、钙结合态磷(Ca-P)以及残渣磷(Res-P), 沉积物无机磷形态分析方法采用Rydin改进的Psenner法, 各形态磷经不同提取剂连续分步提取获得[17, 18]. ①NH4Cl-P(用1mol ·L-1 NH4Cl提取); ②BD-P(用0.11mol ·L-1 Na2S2O4/NaHCO3提取); ③Al-P(用0.1mol ·L-1 NaOH提取); ④Org-P(用0.1mol ·L-1 NaOH提取并消解后除Al-P之外的部分); ⑤Ca-P(用0.5mol ·L-1 HCl提取); ⑥Res-P(沉积物经高温灼烧并用提取得到的TP值减去上述①~⑤步提取磷的总和).各形态磷浓度采用钼酸铵分光光度法测定.从Zr-oxide DGT中取出的Zr-oxide固定膜在混合钼酸盐试剂中35℃着色45 min, 然后利用平板扫描仪获得膜表面着色后的图像, 经过计算机成像密度测定(CID)技术转换成灰度测定DGT不稳定P(DGT-labile P)的浓度[19].
1.5 数据分析实验数据处理及作图采用Origin Pro 2018软件; 数据统计分析采用SPSS 20.0.0软件; 使用方差分析和LSD最小显著性差异法检验实验组之间以及与实验组与对照组之间各特征指标差异的显著性, 显著性水平α=0.05.
依据预先建立的膜表面着色灰度与P积累量(M)之间的定量关系, 得到待处理样品膜上P积累量.根据公式(1)计算不稳定P的通量[20].
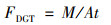
|
(1) |
式中, M为P的累积量, μg; A为固定膜暴露面积, cm2; t为DGT装置在沉积物中的时间, s.
2 结果与讨论 2.1 沉积物中磷固定效果沉积物中注射磷固定剂是为了增大材料与沉积物的接触面积, 利用固定剂的磷吸附性, 使表层沉积物成为一种活性屏障, 既能吸附水体中的营养物质, 又能阻止沉积物中营养物质的释放[21]. 图 1展示了不同方式处理后沉积物间隙水中SRP浓度变化.结果表明, 实验初期24 h内Control组的SRP浓度范围为1.31~3.31 mg ·L-1, 最大值和最小值分别出现在25 mm和0 mm深度处.而注入Ca(OH)2后, 0~20 mm深度的SRP短时间内便降至0.3 mg ·L-1左右, 且整个实验过程中并未有明显增加, 与Control组相比, 添加磷固定剂减少了0~20 mm沉积物中90%以上的磷酸盐. 20~40 mm沉积物中的SRP浓度在短时间内也有明显下降(P<0.05), 但下降效果并没有0~20 mm深度沉积物明显, 与Control组相比, 磷固定剂减少了20~40 mm沉积物中50% ~60%的磷酸盐.而在40 mm以下深度的SRP浓度在注入Ca(OH)2后12h内并未有明显变化(P=0.85), 且在12 h后, 40 mm以下深度SRP浓度发生明显增加, 第144 h时45 mm和55 mm深度SRP浓度分别为2.88 mg ·L-1和3.33 mg ·L-1, 相比于同时期Control组相同深度SRP浓度上升81.6%和82.9%.这一结果表明, 单纯投加磷固定剂仅能对SWI以下约40 mm左右范围进行有效控制, 且控制效果会随着深度的增加而大幅度削弱.而且, 磷固定剂的加入会导致40 mm深度以下SRP浓度明显增加.推测出现这一现象的原因:首先当表层沉积物中磷被有效固定后, 因浓度梯度的影响, 深层沉积物中内源磷会形成向表层沉积物迁移的趋势, 因而磷酸盐会在固磷材料影响较弱的区域富集.此外, 固体材料会在沉积物中形成隔离层, 阻碍氧向深层沉积物中的渗透, 从而导致沉积物微环境的厌氧状态加剧[22], 进一步诱发内源磷释放.

|
图 1 沉积物-水剖面中SRP浓度随时间的变化 Fig. 1 Changes in the concentrations of SRP in porewater over time in sediment-water profiles |
氧化剂和Ca(OH)2以及[OX+Ca(OH)2组]混合投加后, 0~60 mm深度沉积物中SRP浓度都得到了有效控制.与Ca(OH)2组相比, OX+Ca(OH)2组对SWI以下0~20 mm深度的SRP控制效果并不优于Ca(OH)2组, 甚至有时略逊色于Ca(OH)2组, 这主要归因于易释放态磷因氧化作用短期内大量释放[23], 磷固定剂难以完全控制溶解性磷酸盐随间隙水迁移.随着Ca(OH)2固磷效果因沉积物深度的增加而迅速削弱, Ca(OH)2组20 mm以下深度SRP浓度明显高于OX+Ca(OH)2组, 这表明氧化剂的加入的确可以提高磷固定剂在沉积物中的纵向影响力.对于40 mm深度以下沉积物, 相较于对0~40 mm间隙水中SRP 71.89%的控制效果, 其对40 mm以下间隙水中SRP的控制效果仅为35.58%, 这说明OX+Ca(OH)2组对SWI下40 mm以下间隙水控制效果出现了明显削弱.
沉积物DGT-labile P通量的变化如图 2所示, 由DGT-labile P的水平平均通量得到P通量的一维图像, 如图 3所示.经计算, Control组SWI界面下0~48 mm沉积物DGT-labile P通量平均值为99.66pg ·(cm2 ·s)-1, 其于SWI界面下0~20 mm出现陡增, 在20 mm深度处的通量约为121.70pg ·(cm2 ·s)-1, 20 mm深度以下通量增长趋于平缓, 于30 mm深度达到峰值136.77pg ·(cm2 ·s)-1后缓慢降低.单独注入Ca(OH)2后, DGT-labile P通量在SWI界面下0~30 mm显著低于Control组, 其0~20 mm深度DGT-labile P通量仅为Control组同深度通量的10%, 但Ca(OH)2组DGT-labile P通量在SWI界面下20~40 mm出现激增, DGT-labile P通量由SWI界面下20 mm深度的23.18pg ·(cm2 ·s)-1激增至40 mm深度的222.21pg ·(cm2 ·s)-1, 增长858.63%, 于40 mm深度左右达到峰值后迅速降低, 经计算Ca(OH)2组SWI界面下0~48 mm沉积物DGT-labile P通量平均值为86.05pg ·(cm2 ·s)-1; OX+Ca(OH)2组处理后DGT-labile P通量均显著低于Control组, 其SWI界面下0~48 mm沉积物DGT-labile P通量平均值为32.71pg ·(cm2 ·s)-1, 同比于Control组分别降低了67.18%, 而OX+Ca(OH)2组DGT-labile P通量在SWI界面下20~44 mm深度出现迅速增长, 由20 mm处11.19pg ·(cm2 ·s)-1增长至44 mm处60.85pg ·(cm2 ·s)-1, 增长约443.79%.

|
深度0处为SWI界面 图 2 处理后沉积物中二维DGT-labile P通量的变化 Fig. 2 Variations in 2D DGT-labile P flux in the sediment after treatment |

|
深度0处为SWI界面 图 3 处理后沉积物中二维DGT-labile P通量水平平均值的变化 Fig. 3 Variations in the horizontal average 2D DGT-labile P flux in the sediment after treatment |
由此可见, 氧化剂的确可以强化磷固定剂在沉积物中的纵向影响力.而出现这种结果的原因可能有:①首先氧化剂的加入使得沉积物中大量有机质分解, 加速沉积物的矿化, 从而使得固磷材料的影响效果更容易达到深层沉积物[24]; ②其次, 氧化剂使得沉积物中不稳定态磷分解释放, 并立刻被与氧化剂一同注射的Ca(OH)2所固定转化为稳定态磷, 从而达到短时间内固定内源磷的效果; ③另外氧化剂大幅度提高了沉积物中的氧化还原电位, 将还原态的FeS氧化为铁氧(氢氧)化合物(FeOOH), 吸收间隙水中的磷酸盐[25].
2.2 氧化还原环境影响为探究氧化剂是否是利用其氧化性从而促进了磷固定材料的纵向作用深度, 本研究运用微电极技术考察氧化剂对沉积物纵向微环境的影响, 研究对象为氧化剂影响最为显著的ORP.投加氧化剂前后沉积物中ORP变化如图 4所示.

|
图 4 投加氧化剂前后间隙水中ORP的垂直分布 Fig. 4 Vertical profiles of ORP in porewater before and after the addition of the oxidant |
如图 4所示, 在未做任何处理的情况下, ORP在SWI以上保持在281.78 mV左右; 而在SWI以下0~10 mm, ORP随着沉积物深度的增加而迅速降低至9.46 mV; 10 mm深度以下ORP继续降低, 到SWI以下40 mm处, ORP降至2.90 mV.向沉积物中投加氧化剂后, 沉积物中各层ORP都明显提高, SWI以下0~10 mm维持在442.02 mV左右; SWI以下10 mm后ORP出现陡降, 但仍明显高于空白组同层沉积物, 经计算投加氧化剂后SWI以下沉积物氧化还原电位提高了近18.37倍, 此外投加氧化剂后, SWI以下20 mm和35 mm出现峰值, 造成这种现象的具体机制尚不明确, 但这表明氧化剂可以有效影响SWI以下超过40 mm深度沉积物中的氧化还原环境.因此, 对于原位覆盖而言, 氧化剂的加入可以显著改善纵向影响不足的缺陷.
2.3 沉积物中氨氮变化众所周知, NH4+-N作为沉积物中的还原物质, 其在不同深度间隙水中的浓度代表了该深度沉积物的氧化还原条件.根据图 5所示, Control组在约为20 mm深度达到峰值, 这与先前的报道结果相似[26].单独投加Ca(OH)2以后, 除60 mm深度, 各层NH4+-N都有明显增加, 平均值浓度相比于Control组增加了约56.71%, 且随着时间推移, 深层沉积物中NH4+-N浓度增加越发显著, 144 h时Ca(OH)2组NH4+-N平均值浓度比于Control组增加了约112.77%, 这说明单独投加Ca(OH)2会导致沉积物中NH4+-N增加, 这一现象可能与以下原因有关:首先是Ca(OH)2在沉积物中的隔离层阻碍了氧向深层沉积物中的渗透[27]; 其次是Ca(OH)2造成沉积物环境碱性增大, 加速氨氮中的分子氨的转化, 从而加剧厌氧环境[28]. OX+Ca(OH)2组因为加入氧化剂的缘故, 其氨氮浓度明显小于Ca(OH)2组; 值得一提的是, 在投加材料后的48 h内, 相较于Control组, OX+Ca(OH)2组氨氮浓度出现剧烈波动, 48 h后, 30 mm以下NH4+-N浓度便不再被控制.这种现象的出现可能是因为氧化剂在处理前期凭其强氧化性, 可以控制沉积物中NH4+-N, 但随着时间推移, 氧化剂的效果弱化, 其对氨氮的控制效果减弱, 转而Ca(OH)2对NH4+-N的释放效果占据主导.这也间接说明氧化剂可以短时间内影响控制深层沉积物.

|
图 5 沉积物-水剖面中NH4+-N浓度随时间的变化 Fig. 5 Changes in the concentrations of NH4+-N in porewater over time in the sediment-water profiles |
按照磷的迁移能力, 沉积物中磷可以分为弱吸附态磷(NH4Cl-P)、铁结合态磷(BD-P)、铝结合态磷(Al-P)、有机磷(Org-P)、钙结合态磷(Ca-P)和残渣磷(Res-P).根据沉积物中形态磷释放特性的不同, 有研究将弱吸附态磷(NH4Cl-P)、铁结合态磷(BD-P)和有机磷(Org-P)归为潜在活性磷[29], 即在合适的环境条件下易于释放的形态磷.因此对内源磷的控制关键在于对潜在活性磷的控制.
图 6为不同处理后沉积物各深度形态磷分布情况.经计算, Control组各层沉积物中潜在活性磷含量分别为25.22%、25.53%和23.19%.单独投加Ca(OH)2后, 各层沉积物TP并未出现明显变化; 0~20 mm沉积物中NH4Cl-P、Al-P和Org-P都出现明显减少, 其中Org-P的减少最为显著, 其减少了约36.47%, Ca(OH)2处理后Org-P在形态磷中比例由处理前的7.28%减少到处理后的4.63%, 与之相对应, 该层沉积物中Ca-P比例增加了5.90%;而在20~40 mm沉积物中, NH4Cl-P和Org-P出现明显增加, 其分别增加了104.25%和46.32%, 同时该层中相对稳定的Al-P减少了约39.12%, 而Ca-P也下降了约1.83%; 40~60 mm沉积物与Control组基本相似, 唯一不同的是Ca(OH)2组相比较于Control组, NH4Cl-P增加了约78.73%以及Org-P减少了约52.01%;经计算, Ca(OH)2组各层沉积物中潜在活性磷含量分别为21.38%、37.68%和25.27%, 与Control组同层潜在活性磷相比分别变化了-3.84%、12.16%和2.08%.这说明单独投加Ca(OH)2仅可以对表层沉积物中潜在活性磷进行有效固定, 而对于20 mm以下沉积物, 投加Ca(OH)2会促进沉积物中潜在活性磷的增长, 使原本相对稳定的Al-P和Org-P转化为更易释放的NH4Cl-P、BD-P和Org-P.

|
图 6 不同处理后沉积物各深度形态磷分布 Fig. 6 Content of forms of P at different sediment depths and at various times during the different treatments |
投加氧化剂和Ca(OH)2后, OX+Ca(OH)2组0~40 mm沉积物TP出现明显减少, 其减少了约6.09%的TP, 造成这一现象的主要原因是沉积物中潜在活性磷因氧化而短时间内分解释放, 并随着间隙水向上覆水中迁移.因为本研究磷固定剂的投加方式为点注射, 难以短时间内将释放的磷吸收固定; OX+Ca(OH)2组0~20 mm沉积物中, BD-P、Al-P和Org-P分别减少了约24.81%、12.54%和86.53%, 相对应的Ca-P增加了约10.03%; 20~40 mm沉积物与0~20 mm沉积物相似, BD-P、Al-P和Org-P明显减少, 其分别减少了约37.89%、9.05%和50.90%, 不同的是该层除Ca-P增加了约4.50%外, NH4Cl-P也增加了约95.69%;与0~40 mm沉积物不同, OX+Ca(OH)2组40~60 mm沉积物TP比Control组同层沉积物TP增加了约5.11%, 分析该层形态磷分布可知, 该层BD-P、Org-P和Ca-P分别增加了约42.01%、61.80%和3.44%, 而NH4Cl-P和Al-P分别减少了约4.29%和13.32%;经计算, OX+Ca(OH)2组各层沉积物中潜在活性磷含量分别为18.00%、23.41%和29.49%, 与Control组同层潜在活性磷相比分别变化了-7.22%、-2.12%和6.29%.因此可以看出, 投加氧化剂虽然会导致一部分磷随间隙水流入上覆水中, 但其的确可以提高Ca(OH)2的固磷深度.
综上所述, 单独投加Ca(OH)2的影响力仅能有效固定SWI以下约20 mm沉积物中的潜在活性磷, 而20 mm深度以下的沉积物则难以影响到, 甚至会造成深层沉积物中潜在活性磷的积增.投加氧化剂可以有效强化Ca(OH)2对沉积物中潜在活性磷的纵向影响力, 具体体现在:①首先氧化剂有效减少沉积物中的潜在活性磷含量, 同时使得同层沉积物中Ca-P含量明显增加, 因此可以推测, 氧化剂的加入使沉积物中不稳定态磷氧化分解, 从而更易于被磷固定剂直接吸附固定; ②其次与Ca(OH)2组相比, 氧化剂的加入明显增加了各层中Ca-P含量, 这说明氧化剂的确促进了Ca(OH)2在沉积物纵向的扩散.但这种方法对40mm以下沉积物影响效果并不明显, 且Ca(OH)2造成的碱性环境加剧沉积物厌氧环境不利于潜在活性磷的固定.
2.5 指示意义原位固定技术因其廉价且易操作的优势而成为控制富营养化湖泊内源磷负荷的常用方法[30].然而粉末状固磷材料难以影响深层沉积物, 且长期覆盖固磷材料会加剧深层沉积物厌氧环境等问题, 一直以来都限制着原位固定技术的发展.根据本研究实验结果:氧化剂与磷固定剂混合注入沉积物中, 可以显著提高材料对深层沉积物的控制效果, 因此氧化剂与磷固定剂混合投加可以对深层沉积物内源磷的控制具有良好的应用前景.但本研究发现投加氧化剂会导致部分内源磷释放到水体中, 因此该方法可与原位覆盖技术配合形成内源磷复合控制系统, 克服其两者的弊端, 从而达到多方位控制水体内源磷负荷的效果.
3 结论(1) 单独投加Ca(OH)2可以有效固定SWI以下约20 mm沉积物中约90%内源磷, 但同时会加剧20 mm以下沉积物的厌氧环境, 造成潜在活性磷的累积.
(2) 加入氧化剂可以短时间内有效削减沉积物内源磷负荷, 处理后沉积物中DGT-labile P通量减少66.95 pg ·(cm2 ·s)-1, 比单独投加Ca(OH)2提高了12.61 pg ·(cm2 ·s)-1, 并明显提高了Ca(OH)2在沉积物中的纵向扩散深度, 其影响范围可以达到SWI以下0~40 mm.
(3) 氧化剂与磷固定剂混合投加可以有效控制深层内源磷负荷, 但其会导致部分内源磷释放到水体中, 因此该方法可与原位覆盖技术配合形成内源磷复合控制系统, 克服其两者的弊端, 从而达到多方位控制水体内源磷负荷的效果.
(4) 氧化剂与Ca(OH)2的混合投加极大改善了单一投加Ca(OH)2时纵向固定效果不足的弊端, 强化了Ca(OH)2对底泥沉积物的纵向影响.
| [1] | Gubelit Y I, Berezina N A. The causes and consequences of algal blooms: the Cladophora glomerata bloom and the Neva Estuary (eastern Baltic Sea)[J]. Marine Pollution Bulletin, 2010, 61(4-6): 183-188. DOI:10.1016/j.marpolbul.2010.02.013 |
| [2] | Smith V H, Tilman G D, Nekola J C. Eutrophication: impacts of excess nutrient inputs on freshwater, marine, and terrestrial ecosystems[J]. Environmental Pollution, 1999, 100(1-3): 179-196. DOI:10.1016/S0269-7491(99)00091-3 |
| [3] | Stow C A, Cha Y. Are chlorophyll a-total phosphorus correlations useful for inference and prediction?[J]. Environmental Science & Technology, 2013, 47(8): 3768-3773. |
| [4] | Liu T Z, Yuan J J, Dong W Y, et al. Effects on inorganic nitrogen compounds release of contaminated sediment treatment with in situ calcium nitrate injection[J]. Environmental Science and Pollution Research, 2015, 22(2): 1250-1260. DOI:10.1007/s11356-014-3421-7 |
| [5] |
丁玉琴, 李大鹏, 张帅, 等. 镁改性芦苇生物炭控磷效果及其对水体修复[J]. 环境科学, 2020, 41(4): 1692-1699. Ding Y Q, Li D P, Zhang S, et al. Phosphate control effect and water body remediation of magnesium modified reed biochar[J]. Environmental Science, 2020, 41(4): 1692-1699. |
| [6] | Nürnberg G K, Tarvainen M, Ventelä A M, et al. Internal phosphorus load estimation during biomanipulation in a large polymictic and mesotrophic lake[J]. Inland Waters, 2012, 2(3): 147-162. DOI:10.5268/IW-2.3.469 |
| [7] | Malmaeus J M, Rydin E. A time-dynamic phosphorus model for the profundal sediments of Lake Erken, Sweden[J]. Aquatic Sciences, 2006, 68(1): 16-27. DOI:10.1007/s00027-005-0801-6 |
| [8] | Søndergaard M, Jensen J P, Jeppesen E. Role of sediment and internal loading of phosphorus in shallow lakes[J]. Hydrobiologia, 2003, 506-509(1-3): 135-145. DOI:10.1023/B:HYDR.0000008611.12704.dd |
| [9] | Yu J H, Ding S M, Zhong J C, et al. Evaluation of simulated dredging to control internal phosphorus release from sediments: focused on phosphorus transfer and resupply across the sediment-water interface[J]. Science of the Total Environment, 2017, 592: 662-673. DOI:10.1016/j.scitotenv.2017.02.219 |
| [10] |
林建伟, 朱志良, 赵建夫. 曝气复氧对富营养化水体底泥氮磷释放的影响[J]. 生态环境, 2005, 14(6): 812-815. Li J W, Zhu Z L, Zhao J F. Effect of aeration on release of nitrogen and phosphorus from sediments in eutrophic waterbody[J]. Ecology and Environment, 2005, 14(6): 812-815. DOI:10.3969/j.issn.1674-5906.2005.06.002 |
| [11] | Fan Y, Li Y W, Wu D Y, et al. Application of zeolite/hydrous zirconia composite as a novel sediment capping material to immobilize phosphorus[J]. Water Research, 2017, 123: 1-11. DOI:10.1016/j.watres.2017.06.031 |
| [12] | Wang C H, Jiang H L, Yuan N N, et al. Tuning the adsorptive properties of drinking water treatment residue via oxygen-limited heat treatment for environmental recycle[J]. Chemical Engineering Journal, 2016, 284: 571-581. DOI:10.1016/j.cej.2015.09.011 |
| [13] | Wang L H, Li J G, Zhou Q, et al. Rare earth elements activate endocytosis in plant cells[J]. Proceedings of the National Academy of Sciences of the United States of America, 2014, 111(35): 12936-12941. DOI:10.1073/pnas.1413376111 |
| [14] | Lürling M, Waajen G, Van Oosterhout F. Humic substances interfere with phosphate removal by lanthanum modified clay in controlling eutrophication[J]. Water Research, 2014, 54: 78-88. DOI:10.1016/j.watres.2014.01.059 |
| [15] | Wang Y, Ding S M, Wang D, et al. Static layer: A key to immobilization of phosphorus in sediments amended with lanthanum modified bentonite (PhoslockⓇ)[J]. Chemical Engineering Journal, 2017, 325: 49-58. DOI:10.1016/j.cej.2017.05.039 |
| [16] | Sun Q, Lin J, Cao J X, et al. A new method to overall immobilization of phosphorus in sediments through combined application of capping and oxidizing agents[J]. Science of the Total Environment, 2019, 694. DOI:10.1016/j.scitotenv.2019.133770 |
| [17] | Rydin E. Potentially mobile phosphorus in Lake Erken sediment[J]. Water Research, 2000, 34(7): 2037-2042. DOI:10.1016/S0043-1354(99)00375-9 |
| [18] | Psenner R, Pucsko R, Sager M. Fractionation of organic and inorganic phosphorus compounds in lake sediments[J]. An Attempt to Characterize Ecologically Important Fractions, 1984, 70(1): 111-155. |
| [19] | Ding S M, Wang Y, Xu D, et al. Gel-based coloration technique for the submillimeter-scale imaging of labile phosphorus in sediments and soils with diffusive gradients in thin films[J]. Environmental Science & Technology, 2013, 47(14): 7821-7829. |
| [20] | Li C, Ding S M, Yang L Y, et al. Diffusive gradients in thin films: devices, materials and applications[J]. Environmental Chemistry Letters, 2019, 17(2): 801-831. DOI:10.1007/s10311-018-00839-9 |
| [21] | Xu Y, Han F E, Li D P, et al. Transformation of internal sedimentary phosphorus fractions by point injection of CaO2[J]. Chemical Engineering Journal, 2018, 343: 408-415. DOI:10.1016/j.cej.2018.03.028 |
| [22] | Zhou J, Li D P, Chen S T, et al. Sedimentary phosphorus immobilization with the addition of amended calcium peroxide material[J]. Chemical Engineering Journal, 2019, 357: 288-297. DOI:10.1016/j.cej.2018.09.175 |
| [23] | Yamada T M, Sueitt A P E, Beraldo D A S, et al. Calcium nitrate addition to control the internal load of phosphorus from sediments of a tropical eutrophic reservoir: microcosm experiments[J]. Water Research, 2012, 46(19): 6463-6475. DOI:10.1016/j.watres.2012.09.018 |
| [24] | Lin J, Zhong Y F, Fan H, et al. Chemical treatment of contaminated sediment for phosphorus control and subsequent effects on ammonia-oxidizing and ammonia-denitrifying microorganisms and on submerged macrophyte revegetation[J]. Environmental Science and Pollution Research, 2017, 24(1): 1007-1018. DOI:10.1007/s11356-016-7828-1 |
| [25] | Feibicke M. Impact of nitrate addition on phosphorus availability in sediment and water column and on plankton biomass —Experimental field study in the shallow brackish schlei Fjord (Western Baltic, Germany)[J]. Water, Air, and Soil Pollution, 1997, 99(1-4): 445-456. DOI:10.1007/BF02406884 |
| [26] | Inoue T, Sugahara S, Seike Y, et al. Short-term variation in benthic phosphorus transfer due to discontinuous aeration/oxygenation operation[J]. Limnology, 2017, 18(2): 195-207. DOI:10.1007/s10201-016-0501-z |
| [27] | Zhou J, Li D P, Zhao Z H, et al. Phosphorus immobilization by the surface sediments under the capping with new calcium peroxide material[J]. Journal of Cleaner Production, 2020, 247. DOI:10.1016/sssj.jclepro.2019.119135 |
| [28] |
刘海伟, 刘云, 王海云, 等. pH和共存阳离子对草莓茎吸附水体氨氮的影响[J]. 环境科学, 2010, 31(8): 1884-1889. Liu H W, Liu Y, Wang H Y, et al. Effects of pH and coexisting cations on ammonia adsorption from aqueous solution by strawberry stem Powder[J]. Environmental Science, 2010, 31(8): 1884-1889. |
| [29] |
黄清辉, 王磊, 王子健. 中国湖泊水域中磷形态转化及其潜在生态效应研究动态[J]. 湖泊科学, 2006, 18(3): 199-206. Huang Q H, Wang L, Wang Z J. Advance in the study on phosphorus speciation, transformation and its potential ecological effects in Chinese lakes[J]. Journal of Lake Sciences, 2006, 18(3): 199-206. DOI:10.3321/j.issn:1003-5427.2006.03.002 |
| [30] |
徐垚, 李大鹏, 韩菲尔, 等. CaO2不同投加方式对黑臭河道底泥内源磷释放抑制作用[J]. 环境科学, 2017, 38(7): 2836-2842. Xu Y, Li D P, Han F E, et al. Inhibition of internal phosphorus release in the black-odor channel under different adding methods of CaO2[J]. Environmental Science, 2017, 38(7): 2836-2842. |