 2021, Vol. 42
2021, Vol. 42



2. 巴彦淖尔市环境信息监控中心, 巴彦淖尔 015000;
3. 中国科学院大学, 北京 100049;
4. 巴彦淖尔市环境科学研究所, 巴彦淖尔 015000
2. Bayannaoer Environmental Information Monitoring Center, Bayannaoer 015000, China;
3. University of Chinese Academy of Sciences, Beijing 100049, China;
4. Bayannaoer Institute of Environmental Science, Bayannaoer 015000, China
全氟化合物由于其表面活性、耐热性和耐酸性等特点被广泛使用于大量产品中[1], 包括纺织品、消防泡沫和清洗溶液等.然而, PFASs具有难降解和远距离传输等特性, 也使其在土壤、沉积物、水、生物和其它环境介质中被频繁检出[2~4], 由于其具有明显的生态毒理效应, 对生物和环境都存在潜在风险, 因此人们对PFASs的关注日益增加. 2000年以来, 许多国家和国际组织对全氟类化学物质(尤其是PFOA和PFOS)的生产和使用进行了限制, 而我国政策相对宽松, 巨大的市场需求使得PFASs发生了明显的产能转移[5], 氟化工产业在我国得到了迅速扩散和发展, 辽宁、山东和江苏等地都相继建立起了氟化工产业园区, 且各环节均会带来PFASs的排放.内蒙古在产业结构调整过程中, 也积极推进了氟化工系列产品项目对经济发展的拉动效应. 2006年, 内蒙古首家氟化工园区在乌兰察布建起, 次年, 万豪氟化工项目落户乌兰察布丰镇市, 建设规模为年产偏氟乙烯12 000 t、聚偏氟乙烯7 000 t、氟橡胶3 500 t和PFOA 60 t.加之含氟产品在日常使用过程中导致的含氟化合物排放, 内蒙古自然环境遭到了越来越大的PFASs排放威胁.
水体是各类污染源PFASs的重要接纳和传输转运介质, 近几年已有大量研究报道了水体中PFASs的赋存水平与累积特征, 例如我国的辽东湾[6]、莱州湾[7]、海河流域[8]、珠江三角洲[9]和长江三角洲地区[10]都报道检出PFASs, 甚至远离污染源的偏远地区也有发现.随着河流、湖泊及海洋中PFASs含量的增加, 鱼和虾等水生生物体内也检出了PFASs[11], 再经由食物链进入鸟类和海豚等体内, 目前已有研究发现人类血清中检出PFASs[12].然而目前流域尺度上PFASs的研究主要集中在沿海工业活动较多的地区, 内陆以农业生产活动为主的地区还缺乏关注, 目前乌梁素海流域主要关注传统氮磷的研究, 而针对新兴污染物PFASs的研究尚属空白.内蒙古乌梁素海流域内水系复杂, 经由人工干渠引进黄河水, 一系列工农业活动后的排水又由人工排干排入乌梁素海, 它接纳了河套地区90%以上的农田排水[13], 客观上起到改善黄河水质、控制河套地区盐碱化等关键作用, 对减少巴彦淖尔市排水对黄河水质的直接影响具有不可替代的作用.然而随着当地工、农业活动的增加, 流域内水环境质量受到一定影响, 乌梁素海作为当地唯一的农业废水和生活污水的排水渠道, 污染水平呈增高趋势也将对下游生态环境造成影响, 其流域内水环境质量备受关注.
因此, 本研究对乌梁素海流域不同季节地表水中17种全氟化合物进行了调查, 系统分析了研究区地表水中全氟化合物的空间分布特征及时间变化, 揭示了研究区PFASs的来源, 并评价了乌梁素海流域PFASs的潜在生态风险, 以期为乌梁素海流域水环境保护及全氟化合物管控提供决策依据与理论支持.
1 材料与方法 1.1 研究区概况与样品采集乌梁素海(N 40°47′~41°03′, E 108°43′~108°57′, 海拔1 018.79 m)位于内蒙古自治区巴彦淖尔市乌拉特旗北部, 南北长约35~40 km, 东西宽约5~10 km.湖泊每年11月初开始结冰, 次年3月末开始消融, 冰封期约5个月.整个河套灌区2017~2019年以来工农业生产规模及水平没有明显变化, 但灌区工农业活动具有明显的季节性, 10月为当地农业生产及秋季用水高峰期, 而4月为当地农业生产停工期与冰封期, 此外, 降雨量也有明显的季节性差异, 降雨多集中于夏秋季7~10月, 而4月降雨量少蒸发量大, 两个季度具有时间上的可对比性, 因此本研究针对乌梁素海流域水体, 分别于2017年10月(丰水期)和2019年4月(枯水期)选取包括湖区、排干、黄河及干渠在内30个采样点进行了地表水样品采集(图 1), 对所采集水样17种典型PFASs的暴露特征、空间分布及时间变化进行系统分析.

|
图 1 乌梁素海流域采样点位置示意 Fig. 1 Location of sampling sites in the Wuliangsuhai watershed |
本研究所涉及的17种PFASs分别为:全氟丁酸、全氟戊酸、全氟己酸、全氟庚酸、全氟辛酸、全氟壬酸、全氟癸酸、全氟十一酸、全氟十二酸、全氟十三酸、全氟十四酸、全氟十六酸、全氟十八酸、全氟丁基磺酸、全氟己基磺酸、全氟辛基磺酸和全氟癸烷磺酸.另外, 5种PFASs同位素标记物为:PFBA(1, 2, 3, 4 13C)、PFOA(1, 2, 3, 4 13C)、PFNA(1, 2, 3, 4, 5 13C)、PFDA(1, 2 13C)和PFOS(1, 2, 3, 4 13C). PFASs及同位素标记物纯度>98%(Wellington实验室, 加拿大).色谱级甲醇、乙腈和醋酸铵(J.T. Baker公司, 美国).氨水溶液(28%~30% NH3, Sigma公司, 美国).
样品处理分析使用的主要仪器有:ENVI-carb(Supelco公司), SPE小柱(Oasis WAX, Waters公司, 美国).高效液相色谱(Agilent 1200型, 美国)/质谱(SCIEX 3000型, 美国)联用仪(HPLC/MS-MS).
1.3 样品前处理水样采用OasisTM WAX固相萃取柱进行萃取:取400 mL的水样, 加入5 ng内标, 通过由4 mL氨水甲醇溶液(0.10%), 4 mL甲醇, 4 mL蒸馏水依次清洗过的OasisR WAX柱, 保持样液以1滴·s-1的速度过柱.然后用4 mL的醋酸铵缓冲液(25 mmol·L-1, pH=4)冲洗, 待柱内干燥后, 用4 mL甲醇、4 mL的氨水甲醇溶液(0.10%)洗脱待测物质, 洗脱液收集后用高纯氮气吹至1 mL.并用0.2 μm的尼龙滤膜过滤, 收集待上机.
1.4 液相色谱/质谱条件水样目标化合物的定性定量采用HPLC/MS-MS联用仪.色谱条件:流动相为2 mmol·L-1醋酸铵和乙睛, 柱温30℃, 保留时间40 ms, 流速0.3 mL·min-1, 进样量10.0 μL.质谱条件:电喷雾电离负源, 雾化温度450℃, 辅助气(N2)流量5 L·min-1, 毛细管电压3 500 V.
1.5 质量控制及保证样品在采集过程中采用的是聚丙烯材料的塑料袋, 分析过程中严格控制并避免了含氟材料的使用.同时, 所有的实验容器和装置在使用前均用超纯水和甲醇进行了前处理.为保证数据的可靠性, 分析中包括了场地空白、运输空白和程序空白等操作, 所有处理空白均未超过检测限, 分析标准偏差均在允许范围内((20%).内标法定量, 方法的最低检测限(信噪比3:1)为0.01~0.1 ng·L-1, 定量限(信噪比为5:1)为0.04~0.15 ng·L-1, 样品的加标回收率为66.8%~102.3%(表 1).
|
|
表 1 水中17种目标PFASs的检测限、定量限、回收率和检出率 Table 1 Detection limits, quantitation limits, recovery rates, and detection rates of 17 target PFASs in water |
1.6 数据分析
数据分析使用SPSS 25、Excel 2016和Origin 2018等软件, 空间制图使用ArcGIS 10.5软件.本文通过毒性数据中值和环境数据中值的差异对比定量污染物的相对风险:
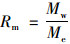
|
(1) |
式中, Rm为某污染物基于中值比较的相对风险值, Mw为该污染物淡水环境浓度中值, Me为该污染物淡水毒性数据中各物种对应数据的中值:

|
(2) |
式中, mi为物种i对应的毒性数据的中值, n为某污染物对应的毒性数据的物种总数, 详细数据来源于团队先前研究工作[14]. Rm值反映了环境数据和毒性数据整体的接近程度, Rm值的大小排序即代表了各污染物相对风险的大小排序[15, 16].利用该方法建立每种物质的生态毒理学数据库和环境暴露数据库, 并根据环境数据中值和毒性数据中值的比或环境数据和毒性数据范围的重叠程度来对每种物质进行相对风险值的定量, 最后分别进行多种污染物的相对风险排序, 可以有效地筛选出水生生物带来最高风险的化学物质, 并能反映出针对不同研究区域和不同风险受体的风险分布格局.
2 结果与讨论 2.1 水体中PFASs的残留水平及组分特征样品PFASs各个组分的质量浓度及贡献百分比(质量分数)分别如图 2和图 3所示.丰水期所检测17种组分中, 羰基碳链小于或等于9的全氟羧酸和PFBS是检出率最高的组分, 检出率都在90%以上, 其中PFBA、PFHxA、PFOA、PFNA和PFBS的检出率达到了100%.各组分总浓度∑PFASs范围4.00~232.05 ng·L-1, 平均值96.01 ng·L-1, 其中PFOA的平均值最高, 达到56.87 ng·L-1, 在∑PFASs中的贡献率高达85%, 远高于检出平均值为第二的PFBA(5.91 ng·L-1, 贡献率9%).

|
图 2 乌梁素海流域水体中PFASs的浓度 Fig. 2 Concentrations of PFASs in the water of the Wuliangsuhai watershed |

|
图 3 乌梁素海流域水体中PFASs的组成 Fig. 3 Composition of PFASs in the water of the Wuliangsuhai watershed |
枯水期的样品中羰基碳链小于或等于8的短链全氟化合物是检出率较高的组分, 检出率都在90%以上, 其中PFBA、PFPeA、PFHxA、PFHpA、PFOA和PFHxS的检出率达到了100%.各组分总浓度∑PFASs范围57.57~263.45 ng·L-1, 平均值145.39 ng·L-1, 是丰水期样品浓度平均值的1.5倍.且此次检出平均值最高的是PFPeA, 平均浓度为31.28 ng·L-1, 组分贡献率为24%, 其次为PFBA(平均值29.26 ng·L-1, 组分贡献率23%)、PFHxA(平均值26.78 ng·L-1, 组分贡献率21%)、PFOA(平均值26.54 ng·L-1, 组分贡献率21%)、PFHpA(平均值7.34 ng·L-1, 组分贡献率6%)、PFBS(平均值4.58 ng·L-1, 组分贡献率4%)和PFHxS(平均值2.3 ng·L-1, 组分贡献率2%), 而其他物质的贡献率均小于1%或接近于0%.
从全国来看, PFOS和PFOA是检出率最高的两种单体, 本研究与我国其他区域地表水体中PFASs的含量相比较来看, 丰水期乌梁素海流域水体的PFOS含量整体上处于较低水平, 与辽东湾、巢湖和官厅水库浓度相当; 枯水期乌梁素海流域水体的PFOS含量有所上升.丰水期总排干下游和乌梁素海湖区水体中的PFOA属于高浓度水平(图 2), 高于巢湖和官厅水库等湖泊, 但仍低于受到氟化工企业严重影响的汤逊湖和小清河流域; 枯水期乌梁素海流域水体的PFOA含量则有所下降.另外, 本研究中PFBA含量则远低于已有报道的武汉汤逊湖和大凌河流域(表 2).
|
|
表 2 不同地区地表水中PFASs含量比较1) Table 2 Comparison of PFASs in the surface water of different regions |
2.2 水体中PFASs时空分布
乌梁素海流域地表水总体上呈现自西南向东北的流向, 主要在南部由黄河引水汇入各干渠对河套地区进行灌溉, 而灌溉退水最终又汇集到北部的总排干, 再流入乌梁素海, 由于这种特殊的灌排方式, 整个研究区地表水中PFASs的含量及组成都呈现出显著的空间差异性(图 4).

|
图 4 乌梁素海流域水体中PFASs分布特征 Fig. 4 Distribution characteristics of PFASs in the water of the Wuliangsuhai watershed |
丰水期样品分析结果显示研究区内水体可以分为两类, 一类是以PFBA为主要组分, 整体浓度较低, 位于黄河、干渠水体及入湖断面的干渠(包括八排干、九排干、W4、W5和W6, 图 3和图 4), 其各组分总浓度∑PFASs范围4.00~21.16 ng·L-1, 平均值8.22 ng·L-1; 另一类是以PFOA为主要组分, 浓度较高的采样点位于总排干中下游水体、总排干入湖断面红圪卜、所有湖区水体和总出湖口乌毛计, 其各组分总浓度∑PFASs范围73.26~232.05 ng·L-1, 平均值146.84 ng·L-1.两类水体∑PFASs平均值相差约17倍.第一类水体中, PFBA在∑PFASs中的平均组分贡献率达51%, PFPeA和PFOA的贡献率分别为15%和11%, PFHxA、PFHpA、PFBS、PFHxS和PFOS的贡献率分别为6%、4%、4%、3%和3%, 而长链(碳链长>8)全氟烷酸和全氟羧酸的贡献率多等于或接近于0%.第二类水体中, PFOA在∑PFASs中的平均组分贡献率达91%, PFBA的贡献率为5%, 而其它全氟烷酸和全氟羧酸的贡献率多等于或接近于0%.各组分总浓度∑PFASs最高的水样位于总排干中下游两个点(总排干2、总排干3)及总排干入湖口红圪卜, 自采样点“总排干2”之前汇入的水体明显含有较高的PFOA, 导致总排干下游及湖区内的PFOA占比增加(图 4).两类水体空间差异的原因一是上游黄河和干渠受到面源农业活动的影响较小, 而下游排干和湖区水承接了主要的农田排水; 二是推断灌区中部存在含全氟化合物废水集中排放的工业活动, 并且直接导致了乌梁素海湖内和退黄河水的PFOA占比突增.
枯水期乌梁素海流域黄河引水中的全氟化合物有了较高的污染水平, 表现为:黄河主干地表水、总干渠和其它干支渠中表层水体各组分总浓度∑PFASs范围为57.57~87.50 ng·L-1, 平均值已达71.70 ng·L-1, 远高于丰水期相应水体(范围4.00~21.16 ng·L-1, 平均值8.22 ng·L-1), 此类水体中4种短链全氟羧酸PFBA、PFPeA、PFHxA和PFHpA是主要组分, 这4种短链全氟羧酸占比在78%~90%之间, 造成两个时期差异的主要原因为枯水期黄河上游来水中PFASs有所增高, 其次枯水期水量明显减小导致稀释作用也相应减弱.随着各类排水进入总排干, 全氟化合物在总排干前段也出现了升高(升高至91.42 ng·L-1).各入湖水体中, 总排干入湖断面红圪卜、八排干入湖断面和九排干入湖断面均表现出明显的PFHxA污染, W5作为长济干渠的入湖口, 空间上位于八排干和九排干之间, 也表现出了较高水平的PFHxA污染, 可能是受到点源污染的影响.受各入湖水体的综合影响, 湖区水体∑PFASs范围110.06~263.45 ng·L-1, 平均值160.01 ng·L-1, 略高于丰水期相应水体(∑PFASs范围73.26~199.61 ng·L-1, 平均值132.46 ng·L-1).湖区北部水体(西羊场以北)中主要PFASs组分的占比分别为PFHxA(35%)、PFOA(22%)、PFPeA(21%)和PFBA(13%), 湖区中南部水体(西羊场以南)中主要PFASs组分的占比为PFOA(41%)、PFBA(26%)、PFPeA(23%)和PFHxA(5%).湖区水体出现南北差异的原因主要是采样时间正值湖水冰封期末期, 湖区南北水利联系减弱, 北部水体受到北部入湖口红圪卜、八排干、九排干和W5的影响较大, 与它们呈现出类似的高PFHxA特征, 而南部水体受到影响较小, 依旧保持丰水期高PFOA的特征.
导致丰水期与枯水期两个时期PFASs含量与组成差异的原因可能包括两点:首先, PFHxA等短链全氟羧酸作为PFOA替代物产量逐渐增加, 导致2019年枯水期黄河水与干渠水样中PFHxA等短链全氟羧酸含量及占比有明显增加, 湖区水体中短链PFASs含量也因干渠水和灌区排水的进入而升高; 其次, 河套灌区每年净引黄水量44×108 m3左右[25], 经一系列农业活动后主要由总排干红圪卜排入乌梁素海, 引黄水量对PFASs浓度有一定的稀释作用, 而引黄入湖水量又具有明显的季节性, 根据2013~2018年巴彦淖尔市水资源公报及总排干红圪卜断面水量监测数据的多年平均值, 丰水期整个河套地区引黄水量较高, 多年平均值为7.63×108 m3; 而枯水期处于当地的冰封期末期, 引黄水量减少, 多年平均值为2.71×108 m3; 稀释作用导致丰水期样品中PFASs相对偏低(图 5).

|
图 5 2013~2018年红圪卜断面入湖水量月平均值及样品中PFASs浓度 Fig. 5 Monthly average of the amounts of water flowing into the lake from 2013 to 2018 at Honggebu and the content of PFASs in the samples |
PFOS/PFOA和PFOA/PFNA常被用于确定PFASs的潜在来源[26, 27].在本研究中, PFOS/PFOA的范围是0~3.18(图 6), 其中枯水期的“总干渠2”和“支渠1”以及丰水期“支渠2”这3个样品PFOS/PFOA大于1, 分别为3.18、1.42和1.17.研究显示污水处理厂来源的PFASs中PFOS/PFOA大多高于1, 例如韩国Shihwa工业区附近的水体, 其PFOS/PFOA大于1.0[17], 另外, 在我国广州和西班牙地中海等地的污水处理厂废水中发现PFOS/PFOA也均大于1.0[28].表明采样点“总干渠2”、“支渠1”和“支渠2”可能受到污水处理厂或氟化工产业点源影响, 因为这3个采样点较靠近巴彦淖尔市区.其余样品PFOS/PFOA都小于1, 这一比值与我国太湖[19]、珠江三角洲地区的5条典型河流相似[17], 这些样品PFASs一方面受到大气远距离传输来源的影响, 另一方面当地存在点源污染造成水体中较高浓度的PFOA, 特别是在工农业活动高峰的丰水期, 总排干及湖区水体中PFOS/PFOA受到点源PFOA排放的影响而远小于1.总体上, 乌梁素海流域内PFASs主要受到点源污染影响, 其次受到大气远距离传输影响.

|
图 6 样品中PFOS/PFOA、PFOA/PFNA及PFHpA/PFOA的比值 Fig. 6 Ratios of PFOS/PFOA, PFOA/PFNA and PFHpA/PFOA in the samples |
乌梁素海流域地表水中PFOA/PFNA的值在2.22~5282.67之间.有研究表明偏远地区例如意大利阿尔卑斯山和南极洲, 其依靠大气传输来源的PFOA/PFNA值分别为1.5±0.8和0.18±0.09[29, 30], 而Armitage等[31]估计1951~2010年制造过程中直接排放的PFOA/PFNA的值为7~15;综合表明了间接来源的PFASs其PFOA/PFNA较小[30](< 7).在本研究区, 采样点“总排干2”上游的地表水样品中PFOA/PFNA值较小(2.22~44.99), 而位于采样点“总排干2”下游及湖区的地表水样品中PFOA/PFNA值突然增高, 范围为49.96~5 282.67, 远高于Armitage等[29]估算的7~15, 表明研究区总排干中下游附近可能存在PFOA的工业输入点源污染造成PFOA的突增.
PFHpA/PFOA的比例可以作为大气沉降的有效示踪剂[32], 通常城市地区PFHpA/PFOA值在0.5~0.9之间, 而偏远地区该值在6~16之间.本研究中乌梁素海流域地表水中PFHpA/PFOA的值在0.01~54.85之间.枯水期样品中黄河水与干渠水的PFHpA/PFOA>6, 这与当地季节性的工农业活动有关, 11月至次年3月为期5个月的冰封期内工农业生产活动频率明显下降, 因此枯水期乌梁素海流域上游的黄河水与干渠水受到排污影响较小; 而其余点位(主要是排干水及湖区水)PFHpA/PFOA大多小于或接近于1, 说明这一部分水体中PFHpA和PFOA的来源主要是城市或工业区的点源污染.
2.4 PFASs对水生生物的潜在影响乌梁素海存在多种污染物复合污染的情况, 为了进行水环境污染的预防与控制, 需要了解水环境污染状况, 确定水环境污染物的风险程度, 筛选优先污染物.因此本文采用风险排序方法来量化PFASs对当地水生生物的威胁程度.本研究选取了重金属、氨氮、PFOA和PFOS等10种污染物进行了相对风险值计算, 其中重金属与氨氮的环境数据值由2019年在研究区所采集的地表水体样品测得, 毒性数据主要来自我国多种淡水物种毒性数据测定值[14, 33].基于毒性数据值和环境监测数据中值比较, 计算得到了10种无机和有机化合物的相对风险值, 最终给出风险排序结果(表 3), 相对风险值的范围为1.72×10-7~0.05, 环境风险指数水平由高到低依次为NH3>Cu>As>Zn>Ni>Cd>Cr>Pb>PFOA>PFOS.
|
|
表 3 乌梁素海流域污染物风险排序 Table 3 Risk ranking of pollutants in the Wuliangsuhai watershed |
风险排序结果总体显示了氨氮、Cu和As为乌梁素海流域风险较高的3种污染物, 在其他研究中也表明乌梁素海流域水体有明显的氨氮超标与砷超标现象[34, 35].其余多数化学物质没有对淡水野生生物带来不可接受的毒性影响, 且PFOA与PFOS的风险值较低, 在当地风险优先值低于重金属与氨氮, 但从风险表征来看2019年枯水期PFOA和PFOS的生态风险较高于2017年丰水期的生态风险, 因此有机物的长期积累效应依然不容忽视.
3 结论(1) 乌梁素海流域内地表水样品PFASs浓度范围4.00~263.45 ng·L-1.样品中羰基碳链小于等于8的全氟化合物检出率较高, 检出率都在90%以上, 丰水期样品中PFOA的平均值最高, 达到56.87 ng·L-1, 贡献率高达85%;枯水期样品中检出平均值最高的是PFPeA, 平均浓度为31.28 ng·L-1, 组分贡献率为24%.
(2) 乌梁素海流域内水体在空间上体现两种主要分布特征, 第一种是以全氟丁酸为主要组分, 总体全氟化合物浓度较低的黄河、干渠水体和入湖断面的干渠水体; 第二种是以全氟辛酸为主要组分, 接纳了河套灌区的工业、农业和生活等废水后, 总体全氟化合物较高的总排干中下游水体、总排干入湖断面红圪卜、湖区水体和出湖口水体.受到短链PFASs产量增加与引黄水量变化的影响, 枯水期水体中全氟化合物含量较丰水期样品高.人类活动是决定PFASs污染空间格局的关键因素.
(3) 乌梁素海流域环境介质中全氟化合物污染状况复杂化, 污染水平受到黄河引水、大气沉降和本地污染源的多重影响.首先, 研究区灌区内PFHpA和PFOA的来源主要是点源污染, 研究区总排干中下游附近可能存在PFOA的工业输入点源污染; 其次, 靠近巴彦淖尔市区的水体, 可能受到污水处理厂影响较大.
(4) 基于淡水水生生物的风险评估表明, 当前PFOA与PFOS的风险值较低, 在当地风险管控优先级低于重金属与氨氮, 但其长期的积累效应依然不容忽视.
| [1] | Giesy J P, Naile J E, Khim J S, J, et al. Aquatic toxicology of perfluorinated chemicals[J]. Reviews of Environmental Contamination and Toxicology, 2010, 202: 1-52. |
| [2] | Olsen G W, Mair D C, Lange C C, et al. Per- and polyfluoroalkyl substances (PFAS) in american red cross adult blood donors, 2000-2015[J]. Environmental Research, 2017, 157: 87-95. DOI:10.1016/j.envres.2017.05.013 |
| [3] | Wang P, Wang T Y, Giesy J P, et al. Perfluorinated compounds in soils from Liaodong bay with concentrated fluorine industry parks in China[J]. Chemosphere, 2013, 91(6): 751-757. DOI:10.1016/j.chemosphere.2013.02.017 |
| [4] | Fang S H, Chen X W, Zhao S Y, et al. Trophic magnification and isomer fractionation of perfluoroalkyl substances in the food web of Taihu lake, China[J]. Environmental Science & Technology, 2014, 48(4): 2173-2182. |
| [5] | Wang T Y, Khim J S, Chen C L, et al. Perfluorinated compounds in surface waters from northern China: comparison to level of industrialization[J]. Environment International, 2012, 42: 37-46. DOI:10.1016/j.envint.2011.03.023 |
| [6] | Ding G H, Xue H H, Yao Z W, et al. Occurrence and distribution of perfluoroalkyl substances (PFASs) in the water dissolved phase and suspended particulate matter of the dalian bay, China[J]. Chemosphere, 2018, 200: 116-123. DOI:10.1016/j.chemosphere.2018.02.093 |
| [7] | Zhao Z, Tang J H, Xie Z Y, et al. Perfluoroalkyl acids (PFAAs) in riverine and coastal sediments of Laizhou bay, north China[J]. Science of the Total Environment, 2013, 447: 415-423. DOI:10.1016/j.scitotenv.2012.12.095 |
| [8] | Li F S, Sun H W, Hao Z N, et al. Perfluorinated compounds in haihe river and dagu drainage canal in Tianjin, China[J]. Chemosphere, 2011, 84(2): 265-271. DOI:10.1016/j.chemosphere.2011.03.060 |
| [9] | Liu B, Zhang H, Xie L, et al. Spatial distribution and partition of perfluoroalkyl acids (PFAAs) in rivers of the Pearl River Delta, southern China[J]. Science of the Total Environment, 2015, 524: 1-7. |
| [10] | Jin Y H, Liu W, Sato I, et al. PFOS and PFOA in environmental and tap water in China[J]. Chemosphere, 2009, 77(5): 605-611. DOI:10.1016/j.chemosphere.2009.08.058 |
| [11] | So M K, Taniyasu S, Yamashita N, et al. Perfluorinated compounds in coastal waters of Hong Kong, south China, and Korea[J]. Environmental Science & Technology, 2004, 38(15): 4056-4063. |
| [12] | Gebbink W A, Van Leeuwen S P J. Environmental contamination and human exposure to PFASs near a fluorochemical production plant: review of historic and current PFOA and GenX contamination in the netherlands[J]. Environment International, 2020, 137. DOI:10.1016/j.envint.2020.105583 |
| [13] | Sun S K, Liu J, Wu P, et al. Comprehensive evaluation of water use in agricultural production: a case study in Hetao irrigation district, China[J]. Journal of Cleaner Production, 2016, 112: 4569-4575. DOI:10.1016/j.jclepro.2015.06.123 |
| [14] | 张梦.淡水生态系统中多种污染物相对风险排序方法及其应用[D].北京: 中国科学院环境生态研究中心, 2017. |
| [15] | Donnachie R L, Johnson A C, Moeckel C, et al. Using risk-ranking of metals to identify which poses the greatest threat to freshwater organisms in the UK[J]. Environmental Pollution, 2014, 194: 17-23. DOI:10.1016/j.envpol.2014.07.008 |
| [16] | Donnachie R L, Johnson A C, Sumpter J P. A rational approach to selecting and ranking some pharmaceuticals of concern for the aquatic environment and their relative importance compared with other chemicals[J]. Environmental Toxicology and Chemistry, 2016, 35(4): 1021-1027. DOI:10.1002/etc.3165 |
| [17] | Pan C G, Ying G G, Liu Y S, et al. Contamination profiles of perfluoroalkyl substances in five typical rivers of the Pearl river delta region, south China[J]. Chemosphere, 2014, 114: 16-25. DOI:10.1016/j.chemosphere.2014.04.005 |
| [18] | Wang P, Lu Y L, Wang T Y, et al. Occurrence and transport of 17 perfluoroalkyl acids in 12 coastal rivers in south Bohai coastal region of China with concentrated fluoropolymer facilities[J]. Environmental Pollution, 2014, 190: 115-122. DOI:10.1016/j.envpol.2014.03.030 |
| [19] | Guo C S, Zhang Y, Zhao X, et al. Distribution, source characterization and inventory of perfluoroalkyl substances in Taihu lake, China[J]. Chemosphere, 2015, 127: 201-207. DOI:10.1016/j.chemosphere.2015.01.053 |
| [20] | Liu W X, He W, Qin N, et al. Temporal-spatial distributions and ecological risks of perfluoroalkyl acids (PFAAs) in the surface water from the fifth-largest freshwater lake in China (lake Chaohu)[J]. Environmental Pollution, 2015, 200: 24-34. DOI:10.1016/j.envpol.2015.01.028 |
| [21] | Wang T Y, Chen C L, Naile J E, et al. Perfluorinated compounds in water, sediment and soil from Guanting reservoir, China[J]. Bulletin of Environmental Contamination and Toxicology, 2011, 87(1). DOI:10.1007/s00128-011-0307-y |
| [22] | Zhou Z, Liang Y, Shi Y L, et al. Occurrence and transport of perfluoroalkyl acids (PFAAs), including short-chain PFAAs in Tangxun lake, China[J]. Environmental Science & Technology, 2013, 47(16): 9249-9257. |
| [23] | Wang P, Lu Y L, Wang T Y, et al. Shifts in production of perfluoroalkyl acids affect emissions and concentrations in the environment of the Xiaoqing river basin, China[J]. Journal of Hazardous Materials, 2016, 307: 55-63. DOI:10.1016/j.jhazmat.2015.12.059 |
| [24] | Wang P, Lu Y L, Wang T Y, et al. Coupled production and emission of short chain perfluoroalkyl acids from a fast developing fluorochemical industry: evidence from yearly and seasonal monitoring in Daling river basin, China[J]. Environmental Pollution, 2016, 218: 1234-1244. DOI:10.1016/j.envpol.2016.08.079 |
| [25] |
马雪鑫, 李畅游, 史小红, 等. 乌梁素海水环境容量分析[J]. 灌溉排水学报, 2019, 38(6): 105-112. Ma X X, Li C Y, Shi X H, et al. Environment capacity of the Wuliangsuhai lake[J]. Journal of Irrigation and Drainage, 2019, 38(6): 105-112. |
| [26] | Cao X H, Wang C C, Lu Y L, et al. Occurrence, sources and health risk of polyfluoroalkyl substances (PFASs) in soil, water and sediment from a drinking water source area[J]. Ecotoxicology and Environmental Safety, 2019, 174: 208-217. DOI:10.1016/j.ecoenv.2019.02.058 |
| [27] | Yang Q Q, Wang S L, Liu W J, et al. Spatial distribution of perfluoroalkyl acids (PFAAs) and their precursors and conversion of precursors in seawater deeply affected by a city in China[J]. Ecotoxicology and Environmental Safety, 2020, 194. DOI:10.1016/j.ecoenv.2020.110404 |
| [28] | Campo J, Masiá A, Picó Y, et al. Distribution and fate of perfluoroalkyl substances in mediterranean spanish sewage treatment plants[J]. Science of the Total Environment, 2014, 472: 912-922. DOI:10.1016/j.scitotenv.2013.11.056 |
| [29] | Cai M H, Yang H Z, Xie Z Y, et al. Per- and polyfluoroalkyl substances in snow, lake, surface runoff water and coastal seawater in Fildes Peninsula, King George Island, Antarctica[J]. Journal of Hazardous Materials, 2012, 209-210: 335-342. DOI:10.1016/j.jhazmat.2012.01.030 |
| [30] | Young C J, Furdui V I, Franklin J, et al. Perfluorinated acids in arctic snow: new evidence for atmospheric formation[J]. Environmental Science & Technology, 2007, 41(10): 3455-3461. |
| [31] | Armitage J M, MacLeod M, Cousins I T. Comparative assessment of the global fate and transport pathways of long-chain perfluorocarboxylic acids (PFCAs) and perfluorocarboxylates (PFCs) emitted from direct sources[J]. Environmental Science & Technology, 2009, 43(15): 5830-5836. |
| [32] | Simcik M F, Dorweiler K J. Ratio of perfluorochemical concentrations as a tracer of atmospheric deposition to surface waters[J]. Environmental Science & Technology, 2005, 39: 8678-8683. |
| [33] |
李奇锋, 吕永龙, 王佩, 等. 基于环境风险排序的流域优先污染物筛选[J]. 环境科学, 2018, 39(10): 4472-4478. Li Q F, Lü Y L, Wang P, et al. Selection of priority contaminants in a watershed using risk ranking methodology[J]. Environmental Science, 2018, 39(10): 4472-4478. |
| [34] |
于瑞宏, 刘廷玺, 许有鹏, 等. 人类活动对乌梁素海湿地环境演变的影响分析[J]. 湖泊科学, 2007, 19(4): 465-472. Yu R H, Liu T X, Xu Y P, et al. The impacts of human activities on the Wuliangsuhai wetland environment[J]. Journal of Lake Sciences, 2007, 19(4): 465-472. DOI:10.3321/j.issn:1003-5427.2007.04.016 |
| [35] | Guo H M, Zhang Y, Xing L N, et al. Spatial variation in arsenic and fluoride concentrations of shallow groundwater from the town of Shahai in the Hetao basin, Inner Mongolia[J]. Applied Geochemistry, 2012, 27(11): 2187-2196. DOI:10.1016/j.apgeochem.2012.01.016 |