 2020, Vol. 41
2020, Vol. 41



2. 南京信息工程大学气象灾害预报预警与评估协同创新中心, 中国气象局气溶胶-云-降水重点开放实验室, 南京 210044
2. Key Laboratory for Aerosol-Cloud-Precipitation of China Meteorological Administration, Collaborative Innovation Center on Forecast and Evaluation of Meteorological Disasters, Nanjing University of Information Science&Technology, Nanjing 210044, China
气溶胶新粒子生成(new particle formation, NPF)作为气溶胶的重要来源之一, 对于区域空气污染和全球气候变化具有十分重要的意义[1~4].气溶胶新粒子是指大气中的凝结蒸汽形成的稳定分子簇, 通过碰并或者凝结过程长大至可观测到的粒子[5, 6].这些新生成的粒子在边界层中的增长速率为1~20nm·h-1, 因此经过十几小时的持续增长后, 可对大气云凝结核和城市霾污染产生较大影响[6~9].
NPF事件的研究最早可追溯到1897年, 由Aitken[10]首先推测得出, 但是由于受到观测技术的限制, 当时并没有直接的观测证据.随着气溶胶粒径谱分布观测技术的迅速发展, 目前已在全球不同大气环境中均观测到大量的NPF事件, 例如极地地区、高山、森林、城市地区、乡村地区和沿海地区等[1, 11~14].以往的研究表明NPF事件多发生在相对清洁的地区, 在污染地区, 由于大气背景气溶胶浓度较高, 因此很难观测NPF事件.但是, 近年来在中国地区的大量观测表明即使是在污染大气中, 仍然可观测到较高频率的NPF事件.在气溶胶污染比较严重的京津冀、长三角和珠三角地区均观测到大量的NPF事件[15~27]. Guo等[7]的研究发现在我国城市地区大气复合污染条件下, 由于机动车排放的大量的高浓度挥发性有机物使得环境大气具有极强的新粒子生成潜势, 使得大气中超细颗粒物浓度急剧增加, 进而导致城市地区霾的形成. Nie等[26]则在沙尘期间观测到NPF事件, 发现在沙尘过程中15~50 nm的气溶胶的生成速率和增长速率都有明显的增强.这表明不同大气环境下NPF事件的形成条件和机制存在较大的差异, 因此针对不同大气环境下的观测有利于加深对NPF事件的形成机制的认识.
目前, 中国针对NPF事件的研究多集中在中东部地区和沿海地区, 如北京[14, 15, 28]、青岛[29, 30]、南京[31, 32]、上海[20, 22]和香港[33, 34]等城市地区, 此外在泰山[13]、衡山[26]和黄山[27]等高山站点也有一些观测研究.但是对于内陆城市地区的观测相对较少.内蒙古高原作为中国第2大高原, 平均海拔在1 000~1 200 m, 北连蒙古大戈壁, 南临黄土高原和华北平原.鄂尔多斯位于内蒙古自治区西南部, 地处鄂尔多斯高原腹地, 位于蒙古高原和黄土高原中间位置, 为典型的北温带半干旱大陆性气候区, 全年多盛行西风及北偏西风, 为华北平原的上风向.本研究使用宽范围粒径谱仪于2019年8月16至10月4日在鄂尔多斯观测了10 nm~10 μm气溶胶粒径分布, 结合同期的PM(PM2.5和PM10)、污染气体、气象数据和HYSPLIT模式, 分析了NPF过程中气溶胶粒径分布特征和形成条件, 并根据气团来向分为3类NPF事件, 分析了不同类型NPF过程的气溶胶分布特征和形成条件差异.
1 材料与方法 1.1 观测站点介绍观测点位于鄂尔多斯气象局(39.37°N, 109.49°E; 1 300 m), 位于鄂尔多斯市中南部的康巴什区(图 1), 地处鄂尔多斯高原腹地, 距东胜区25 km、阿勒腾席热镇3 km.康巴什区是鄂尔多斯新的政治文化中心、金融中心、科研教育中心和装备制造基地、轿车制造业基地, 也是全国首个以城市景观命名的4A级旅游景区.观测点北部紧邻考考什纳滨湖公园, 南部紧邻康巴什新区第一小学, 西部为康巴什第一中学, 东部为居民区, 周边无明显排放源.

|
图 1 研究区地形和站点周边分布示意 Fig. 1 Topography of the observation site and the surrounding area |
气溶胶数浓度观测仪器使用美国MSP公司生产的宽范围颗粒粒径谱仪(WPS), 测量粒径范围为0.01~10 μm.关于仪器原理的具体介绍可参见文献[27].观测时间为2019年8月16日至10月4日, 观测时间分辨率设置为5min, 剔除无效和错误数据共获得12 206组有效数据.
气象数据(温度、气压、风速、风向、RH、降水量和能见度)来自鄂尔多斯市康巴什气象观测场, 数据质量控制参照中国气象局气象数据观测标准, 时间分辨率为1 h. PM2.5、PM10、O3、SO2、CO和NO2数据来自中国环境监测总站全国城市空气质量实时发布平台(http://106.37.208.233:20035/)公布的数据, 时间分辨率为1 h.选取距离观测点最近的康泽苑国控点数据, 距离鄂尔多斯市气象局约5.8 km.
1.3 HYSPLIT模式本文所采用的后向轨迹模式是由美国国家海洋及大气管理局(NOAA)开发的轨迹计算模式HYSPLIT(http://ready.arl.noaa.gov/HYSPLIT.php).该模式目前已在大气污染物输送研究中得到了广泛的应用[35].利用HYSPLIT模式计算了观测期间每小时距地500 m高度的24 h后向轨迹.模式采用的气象场为美国国家环境预报中心(NECP)的再分析资料, 水平分辨率为1.0°×1.0°.
1.4 新粒子生成事件判断标准根据以往研究成果[1, 6, 8, 12, 28, 31], NPF过程必须满足以下3个条件:①NPF事件发生时, 核模态气溶胶(10~20 nm)数浓度出现明显的新的峰值, 且持续时间在1 h以上; ②新出现的核模态气溶胶的粒径在大气中持续增长, 在几小时内可增长至爱根核模态(20~100 nm); ③这个新出现的核模态气溶胶峰值与交通源、居民生活源等能源使用过程产生的SO2、CO和NO2等污染气体的浓度无关, 在核模态气溶胶新峰值出现时, 其他粒径段气溶胶没有同时出现峰值.
1.5 生成速率和增长速率计算NPF过程气溶胶粒子的增长速率根据Herrmann等[31]和Kulmala等[36]介绍的方法计算:

|
(1) |
式中, Dp1和Dp2分别是t1和t2的核模态气溶胶粒子的几何中值粒径.
NPF过程气溶胶粒子的生成速率采用了Kulmala等[1]介绍的公式计算:

|
(2) |
式中, Nnuc是核模态气溶胶数浓度, FCoag是由于碰并过程引起的核模态气溶胶的损失通量, Fgrowth表示气溶胶长大超过20 nm上限的通量, 该因子一般可忽略[37].
FCoag可用以下公式计算:
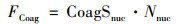
|
(3) |
式中, CoagSnuc是核模态气溶胶的碰并汇.碰并汇的大小直接反映大气中已存在气溶胶对成核新形成气溶胶的碰并去除能力与速率. CoagSnuc可由以下公式计算:
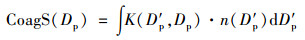
|
(4) |
式中, K(D′p, Dp)是直径为D′p和Dp气溶胶粒子的碰并效率[38].
2 结果与讨论 2.1 新粒子天粒径分布特征观测期间一共出现19次NPF过程, NPF天占总观测天数的37.5%.由图 2可发现, NPF过程中对气溶胶粒径分布的影响较大.一方面, 在NPF过程中气溶胶数浓度, 尤其是100 nm以下的数浓度急剧增加.另一方面, NPF过程中气溶胶的峰值粒径明显增大.虽然NPF生成的气溶胶粒子为20 nm以下的超细粒子, 但是由图 2可发现30~60 nm的气溶胶数浓度在NPF天显著增加.有研究表明, 新粒子生成是云凝结核(CCN)的重要来源, NPF发生后CCN数浓度会明显增加[8].鄂尔多斯平均海拔为1 300~1 500 m, 与中东部地区的边界层顶位置接近, 因此NPF的频发可能会对下游地区的云凝结核分布造成较大影响.

|
图 2 观测期间NPF天和non-NPF天气溶胶粒径分布特征 Fig. 2 Size distribution of number concentration during NPF days and non-NPF days |
由图 2可发现在夜间NPF和non-NPF天气溶胶粒径分布类似, 主要集中在30~80 nm.但是在NPF天, 当新粒子生成事件开始发生后, 10~20 nm的数浓度迅速增加, 在短短的3 h内数浓度增加了5.6倍, 最高可达5 575 cm-3.随后新粒子开始增长, 由图 2(a)可发现: 20~30 nm、30~40 nm和40~50 nm气溶胶数浓度依次开始增加.随着粒径的增大, 数浓度的增长速率逐渐降低, 主要因为气溶胶粒径越大, 所消耗的前体物则越多, 因此增长速率越慢.
此外, 由图 2可发现, 在NPF天和non-NPF天的夜间10 nm附近气溶胶数浓度多观测到高值, 在non-NPF天甚至全天都可观测到10 nm附近的气溶胶数浓度高值.从中可知这些纳米级的气溶胶粒子数浓度高值的持续时间较短, 且没有观测到气溶胶粒子的增长特性, 因此可能主要来自污染源的直接排放.而由图 1可知观测点距离G210和G65等高速公路较近, 且气象局大楼也紧邻伊克昭街, 因此受交通源等的影响较大.此外, 图 2为NPF天和non-NPF天的平均浓度, 可能是由于在某些观测日受到周边排放源的影响, 进而在其中显现出如此分布.
由图 3可发现NPF过程对不同模态气溶胶数浓度日变化的影响不同. NPF事件使得鄂尔多斯市核模态(10~20 nm)和爱根核模态(20~100 nm)气溶胶数浓度急剧增加, 但是对积聚模态(100~1 000 nm)和粗模态(1.0~10 μm)气溶胶数浓度的影响较小. NPF天, 核模态粒子在09:00左右开始急剧增加, 12:00左右达到峰值, 而爱根核模态粒子则在11:30左右才开始快速增加, 并在13:00达到峰值.此后爱根核模态粒子数浓度一直到16:00均维持在较高的浓度, 而此时核模态粒子数浓度则开始快速降低.一方面是因为午后新粒子生成强度减弱, 新生成的粒子减少.另一方面是因为随着核模态粒子的增长, 爱根核模态粒子数浓度增加, 进而对核模态粒子的碰并清除作用也增强.在non-NPF天, 核模态粒子和爱根核模态粒子均没有显著的日变化特征, 但是核模态数浓度在白天要高于夜晚, 而爱根核模态粒子数浓度则是在夜晚高于白天.

|
图 3 观测期间NPF和非NPF不同模态日变化 Fig. 3 Diurnal variations in number concentration in different modes during the NPF days and non-NPF days |
由图 3(c)和图 2(d)可发现NPF天积聚模态和粗模态粒子数浓度要低于non-NPF天.积聚模态粒子数浓度在NPF天和non-NPF天日变化均为单峰型分布, 峰值位于07:00~09:00, 主要受到上班早高峰的影响.粗模态粒子数浓度日变化在NPF天与积聚模态类似, 为单峰型分布; 在non-NPF天为双峰型分布, 在早晚高峰期各有一峰值.
表 1给出了不同地区NPF过程的核模态数浓度、生成速率和增长速率.从中可知, 鄂尔多斯NPF过程中核模态粒子数浓度与南京基本一致, 但是略高于泰山地区.鄂尔多斯市与泰山海拔类似, 泰山位于鄂尔多斯市下游, 这也说明鄂尔多斯市在NPF过程中气溶胶数浓度增加后通过大气环流传输, 进而会对下游地区气溶胶数浓度造成影响.但是由表 1可发现, 鄂尔多斯市NPF过程中核模态粒子数浓度远低于沿海城市嘉兴, 嘉兴位于长三角城市群中心位置, 气溶胶数浓度较高.此外, 嘉兴海拔较低, 而鄂尔多斯市海拔较高接近长三角地区的边界层顶位置, 这也是两地核模态粒子数浓度差异的重要原因.由表 1可知鄂尔多斯市NPF过程中生成速率和增长速率的分布范围与其他地区类似, 这也与Kulmala等[1]总结的全球边界层内NPF过程的生成速率与增长速率相符合.
|
|
表 1 不同地区NPF过程中核模态数浓度、生成速率和增长速率对比1) Table 1 Nucleation number concentration, formation speed and growth speed on NPF events in different cities |
2.2 新粒子天影响因素
由图 4可知PM2.5、PM10和污染气体在NPF天和non-NPF天的日变化特征类似. NPF天PM(PM2.5和PM10)和CO的浓度要远低于non-NPF天.由图 4(a)可发现PM2.5在10:00~11:00出现峰值, 之后浓度迅速下降. PM2.5浓度在NPF天11:00出现最高浓度为14.7 μg·m-3, 在non-NPF天10:00出现最高浓度25.2 μg·m-3. PM2.5在non-NPF天17:00出现最低值为13.9 μg·m-3, 在NPF天则是19:00出现最低值为6.4 μg·m-3. PM10的日变化特征与PM2.5类似. NPF天较低的PM浓度, 意味着大气中气溶胶对太阳辐射的消光作用较弱. NPF天平均能见度和总辐照度分别为34.7 km和471.9W·m-2, 是non-NPF天的1.2和1.5倍.由图 5(f)可发现, NPF天总辐照度要高于non-NPF天, NPF天总辐照度最高为819.9W·m-2, 是non-NPF天的1.4倍.这表明在NPF天到达地面的太阳辐射较高, 有利于光化学反应的发生.由图 4(e)可发现, NPF天11:00~17:00的O3浓度要略高于non-NPF天, NPF天O3最高可达135.6 μg·m-3.

|
图 4 观测期间NPF天和non-NPF天PM2.5、PM10和污染气体日变化 Fig. 4 Diurnal variations in PM2.5, PM10, and pollutant gases during NPF days and non-NPF days |

|
图 5 观测期间NPF天和non-NPF气象要素日变化 Fig. 5 Diurnal variations in meteorological elements during NPF days and non-NPF days |
SO2往往被认为是新粒子生成过程中非常重要的气体组分[41, 42].由图 4(c)可发现SO2在NPF天和non-NPF天均为单峰型分布, 峰值位于09:00~12:00. NPF天SO2浓度在10:00~19:00要高于non-NPF天浓度, 该时间段正好涵盖了新粒子生成过程.在NPF天, 太阳辐射剧烈, 大气氧化性较强, SO2提供了充足的气态H2SO4, 进而促进了新粒子的生成.
NO2主要来自汽车尾气[43, 44].由图 4(d)可发现, NPF天和non-NPF天NO2日变化均为双峰型分布, 早晚高峰的浓度基本类似.但是NPF天的晚高峰时间要比non-NPF天滞后1h. CO则主要来自含碳物质的不完全燃烧, 主要是化石燃料和生物质的燃烧[45].由图 4(f)可知CO的日变化与NO2类似, 说明鄂尔多斯夏秋季CO和NO2主要来自汽车尾气的排放.但是在NPF天和non-NPF天CO的差异要比NO2大. CO作为不完全燃烧的示踪物, 除来自汽车尾气外, 工业燃煤和生物质燃烧排放也是其重要来源[46].由图 6可知在non-NPF天, 主要是偏南气团为主, 气团多经过榆林、延安、临汾和吕梁等地区, 这些地区污染较重.因此NPF天和non-NPF天CO和PM的差异较大. CO虽然也可参与近地面的光化学过程, 是近地面O3的前体物[47], 但是其活性要低于NOx, 因此对NPF天光化学的影响较小.

|
图 6 观测期间500 m高度24 h后向轨迹 Fig. 6 The 24 h back trajectories at an altitude of 500 m during the observation period |
由图 5可发现NPF发生时往往温度较高, 风速较大, 总辐射度较高, RH较低.在日出后, 温度迅速升高, NPF天在07:00时温度为10.8℃, 11:00时温度即升高为21.6℃.总辐照度也迅速增加, 由07:00的116.9W·m-2增加为11:00的598.2W·m-2.高温高辐照度意味着光化学反应剧烈, 因此有利于新粒子的生成.在NPF发生过程中风速较大, 在08:00风速为1.2m·s-1, 13:00达到最大为4.3m·s-1.较高的风速意味着大气扩散条件较好, 因此背景气溶胶数浓度较低, 有利于观测到新粒子生成事件. NPF天RH的日变化要比non-NPF天剧烈, NPF天RH在15:00达到最低26.2%, 仅是non-NPF天的51.2%. NPF天和non-NPF天能见度较高, 尤其是NPF天能见度日变化较小, 均在33 km以上.
由图 6可知, 观测期间鄂尔多斯气团主要分为两大类: 1类为偏北气团, 可占总气团的40.0%;另1类为偏南气团, 可占总气团的54.0%. NPF天共有3类气团类型, 1类是偏西气团, 共观测到3次NPF过程, 占总NPF过程的15.8%.此类气团主要来自鄂尔多斯的西部, 如石嘴山和乌海等地, 为典型的半干旱地区.第2类为偏北气团, 共观测到8次NPF过程, 占总NPF过程的42.1%, 占总偏北气团的40.0%.此类气团多经过鄂尔多斯北部的包头, 甚至来自境外远距离输送.第3类为偏南气团, 也观测到8次NPF过程, 占总偏南气团的29.6%.此类气团多来自鄂尔多斯南部的吕梁、榆林、延安和临汾等地区, 甚至还有来自西安的远距离输送.
由图 6可知NPF天的3类气团的途经地存在显著差异, 这些地区的能源结构、气候和污染源分布特征不同, 因此这些气团的性质也存在较大差异.下文将NPF天按照气团来向分为3类进行讨论.
2.3 不同类型新粒子生成特征由图 7可发现3类NPF过程数浓度粒径分布差异较大.南部气团类型NPF天夜间气溶胶粒径分布更加集中, 多分布在30~60 nm.但是当NPF事件发生前几小时, 气溶胶数浓度显著增加, 且高浓度粒径范围更大, 集中在40~200 nm.由图 7(a)可发现, 在09:30南部气团类型下的NPF过程开始发生, 此时受到早高峰的影响50~100 nm气溶胶数浓度仍然较高, 即南部气团NPF过程发生时背景气溶胶数浓度较高.当NPF过程发生后, 可发现南部气团类型下的气溶胶数浓度迅速增加, 并且迅速向大粒径段增长, 但是在16:00之后NPF过程对气溶胶粒径分布的影响迅速减弱.

|
图 7 3类NPF事件数浓度时间序列 Fig. 7 Size distribution of number concentration for three types of NPF events |
西部气团类型NPF过程对气溶胶粒径分布的影响最小.由图 7(b)可发现在NPF过程发生前气溶胶数浓度变化不大, 在06:00~09:00背景气溶胶浓度略微增大.西部气团类型NPF过程的发生时间较晚, 在11:30才观测到显著的新粒子生成过程. NPF过程的影响时间也较短, 15:00之后影响逐渐消失.
北部气团类型NPF过程的持续时间最长, 由图 7(c)可发现直到20:00之后, NPF事件对气溶胶粒径分布的影响才开始减弱.此外, 在北部气团类型NPF天, 早高峰期间气溶胶数浓度有显著降低, 主要集中在40~60 nm.由图 6可知北部气团类型NPF天气团轨迹较长, 气团的移动速度较快, 大气扩散条件较好.此外, 北部气团多来自相对清洁地区, 气团比较洁净, 因此造成在北部气团类型NPF天的早高峰期间气溶胶数浓度比其他气团类型下数浓度低.尤其是在NPF过程发生前的1 h内, 背景气溶胶显著降低, 在08:30~09:30背景气溶胶在10~100 nm分布比较均匀, 数浓度非常低.但是在NPF开始发生后的2~3 h内, 并没有像南部气团类型NPF过程观测那样观测到显著的粒径增长现象, 而是在11:30之后才出现气溶胶粒子粒径的显著增长过程.
由图 8(a)可发现北部气团类型和南部气团类型下NPF过程中核模态数浓度峰值较高, 明显高于西部气团.由表 2可知北部气团类型和南部气团类型NPF过程中SO2、NO2和CO的浓度要高于西部气团类型NPF过程.北部气团和南部气团中前体污染气体浓度较高, 因此有利于新粒子的形成.北部气团类型NPF天核模态气溶胶数浓度在08:30开始增加, 10:00达到峰值, 此后一直到13:00数浓度才开始减弱.而南部气团类型NPF天核模态气溶胶在达到峰值后, 高浓度仅维持了1 h左右, 就开始显著降低.西部气团类型NPF过程虽然开始较晚, 但是核模态气溶胶高数浓度依然维持了较长时间后才开始降低.此外, 西部气团类型NPF天核模态数浓度虽然在09:40左右就开始观测到显著增加, 但是高浓度仅维持了不到1 h就开始迅速下降, 而此时爱根核模态数浓度反而是下降的趋势, 因此这一时间段并不能归为NPF事件.

|
图 8 不同类型NPF天不同模态气溶胶数浓度日变化 Fig. 8 Diurnal variations in number concentration in different modes during different types of NPF days |
|
|
表 2 不同气团类型NPF事件中气象要素、PM、污染气体、生成速率和增长速率汇总 Table 2 Summary of meteorological elements, PM, pollutant gases, formation rate, and growth rate during different types of NPF events |
由图 7可发现虽然北部气团类型NPF过程发生时间最早, 比南部气团类型NPF过程早了差不多2 h, 但是爱根核模态气溶胶数浓度变化仅比南部气团类型提前了不到1 h.由表 2可知北部气团类型NPF过程增长速率为(10.7±11.1)nm·h-1, 要低于南部气团的(12.7±13.6)nm·h-1.但是北部气团类型NPF过程对气溶胶粒径分布的影响持续时间较长, 由图 7(c)可发现直到21:00其爱根核模态数浓度才恢复到背景浓度, 比南部气团类型晚了接近3 h.
北部气团类型NPF天积聚模态和粗模态气溶胶数浓度较低, 且日变化特征不明显.而西部气团类型和南部气团类型NPF天积聚模态和粗模态气溶胶数浓度均在07:00~10:00观测到显著的峰值.这说明在NPF过程发生前背景气溶胶数浓度的高低并非是决定新粒子生成事件是否发生的关键因素, 但是会对新粒子生成事件的发生时间造成较大影响.
由表 2可知, 不同气团类型NPF过程中气象要素存在显著差异.南部气团类型NPF过程发生时温度最低, 平均为(18.1±2.5)℃; 风速最小, 平均为(2.4±1.5)m·s-1; RH最高, 平均为(48.8±10.8)%; 总辐照度最低, 平均为(447.8±245.2)W·m-2.北部气团类型NPF过程发生时温度最高, 平均为(21.8±2.2)℃; 风速最大, 平均为(4.2±1.9)m·s-1; 总辐照度最高, 平均为(664.5±255.6)W·m-2.西部气团类型NPF过程中RH最低, 平均为(29.8±12.7)%; 气压最高, 平均为(870.9±1.3)hPa.
南部气团类型NPF过程中污染物浓度最高, PM2.5、PM10和NO2的平均浓度分别为(16.8±10.3)、(41.7±19.9)和(20.7±8.6) μg·m-3, 分别是北部气团类型的2.9、1.9和2.0倍, 分别是西部气团类型的1.8、1.6和2.4倍.南部气团类型NPF过程中SO2和CO的浓度略高于北部气团类型, 平均浓度分别为(10.4±5.3) μg·m-3和(0.7±0.1)mg·m-3, 分别是西部气团类型的1.4和1.8倍.这主要是由于气团来源及途经地域所决定.但是西部气团类型NPF过程中O3的浓度最高, 平均浓度为(109.8±23.9) μg·m-3.
不同气团类型NPF过程中新粒子的生成速率相差不大, 西部气团类型最低为(1.5±1.6)cm-3·s-1, 南部气团类型最高为(1.8±1.7)cm-3·s-1.不同气团类型NPF过程中气溶胶的增长速率差异较大, 南部气团类型最大, 为(12.7±13.6)nm·h-1, 是北部气团类型和西部气团类型的1.2倍和1.4倍.南部气团类型NPF过程由于前体气体浓度较高, 风速较低, 有利于新粒子在形成之后的快速增加.而西部气团类型NPF过程中前体气体浓度较低, 虽然O3浓度最高, 光化学过程反应比较剧烈, 因此虽然新粒子生成速率较快, 但是新粒子生成后在大气中的增长速度较慢.
3 结论(1) 观测期间一共出现19次NPF过程, 占总观测期间的37.5%. NPF过程对不同模态气溶胶数浓度日变化的影响不同. NPF过程使得核模态和爱根核模态气溶胶数浓度急剧增加, 但是对积聚模态和粗模态气溶胶数浓度的影响较小.
(2) NPF过程发生时往往温度较高, 风速较大, 总辐照度较高, RH较低. PM2.5、PM10和污染气体在NPF和non-NPF天的日变化特征类似, 但是NPF天PM(PM2.5和PM10)、CO和NO2的浓度较低, O3和SO2较高.鄂尔多斯市夏秋季主导气团主要分为2大类, 1类为偏北气团, 可占总气团的40.0%;另1类为偏南气团, 可占总气团的54.0%. 40.0%的偏北气团和29.6%的偏南气团可观测到NPF事件.
(3) 不同气团类型NPF过程中气象要素存在显著差异.南部气团类型NPF过程发生时风速最小, 平均为(2.4±1.5)m·s-1; RH最高, 平均为(48.8±10.8)%; 总辐照度最低, 平均为(447.8±245.2)W·m-2.北部气团类型NPF过程发生时风速最大, 平均为(4.2±1.9)m·s-1; 总辐照度最高, 平均为(664.5±255.6)W·m-2.西部气团类型NPF过程中RH最低, 平均为(29.8±12.7)%.
(4) 不同气团类型NPF过程中新粒子的生成速率相差不大, 西部气团类型最低为(1.5±1.6)cm-3·s-1, 南部气团类型最高为(1.8±1.7)cm-3·s-1.不同气团类型NPF过程中气溶胶的增长速率差异较大, 南部气团类型最大, 为(12.7±13.6)nm·h-1, 是北部气团类型和西部气团类型的1.2倍和1.4倍.
| [1] | Kulmala M, Vehkamäki H, Petäjä T, et al. Formation and growth rates of ultrafine atmospheric particles:a review of observations[J]. Journal of Aerosol Science, 2004, 35(2): 143-176. DOI:10.1016/j.jaerosci.2003.10.003 |
| [2] | Guo S, Hu M, Zamora M L, et al. Elucidating severe urban haze formation in China[J]. Proceedings of the National Academy of Sciences of the United States of America, 2014, 111(49): 17373-17378. DOI:10.1073/pnas.1419604111 |
| [3] | Lee S H, Gordon H, Yu H, et al. New particle formation in the atmosphere:From molecular clusters to global climate[J]. Journal of Geophysical Research:Atmospheres, 2019, 124(13): 7098-7146. DOI:10.1029/2018JD029356 |
| [4] |
胡敏, 尚冬杰, 郭松, 等. 大气复合污染条件下新粒子生成和增长机制及其环境影响[J]. 化学学报, 2016, 74(5): 385-391. Hu M, Shang D J, Guo S, et al. Mechanism of new particle formation and growth as well as environmental effects under complex air pollution in China[J]. Acta Chimica Sinica, 2016, 74(5): 385-391. |
| [5] | Kulmala M. Atmospheric science:how particles nucleate and grow[J]. Science, 2003, 302(5647): 1000-1001. DOI:10.1126/science.1090848 |
| [6] | Kulmala M, Kontkanen J, Junninen H, et al. Direct observations of atmospheric aerosol nucleation[J]. Science, 2013, 339(6122): 943-946. DOI:10.1126/science.1227385 |
| [7] | Guo S, Hu M, Peng J F, et al. Remarkable nucleation and growth of ultrafine particles from vehicular exhaust[J]. Proceedings of the National Academy of Sciences of the United States of America, 2020, 117(7): 3427-3432. DOI:10.1073/pnas.1916366117 |
| [8] | Yue D L, Hu M, Zhang R Y, et al. Potential contribution of new particle formation to cloud condensation nuclei in Beijing[J]. Atmospheric Environment, 2011, 45(33): 6070-6077. DOI:10.1016/j.atmosenv.2011.07.037 |
| [9] | Williamson C J, Kupc A, Axisa D, et al. A large source of cloud condensation nuclei from new particle formation in the tropics[J]. Nature, 2019, 574(7778): 399-403. DOI:10.1038/s41586-019-1638-9 |
| [10] | Aitken J. Ⅲ.-On some nuclei of cloudy condensation[J]. Earth and Environmental Science Transactions of the Royal Society of Edinburgh, 1900, 39(1): 15-25. DOI:10.1017/S0080456800034025 |
| [11] | Clarke A D, Kapustin V N, Eisele F L, et al. Particle production near marine clouds:Sulfuric acid and predictions from classical binary nucleation[J]. Geophysical Research Letters, 1999, 26(16): 2425-2428. DOI:10.1029/1999GL900438 |
| [12] | Kerminen V M, Chen X M, Vakkari V, et al. Atmospheric new particle formation and growth:review of field observations[J]. Environmental Research Letters, 2018, 13(10): 103003. DOI:10.1088/1748-9326/aadf3c |
| [13] | Shen L J, Wang H L, Yin Y, et al. Observation of atmospheric new particle growth events at the summit of mountain Tai (1534 m) in Central East China[J]. Atmospheric Environment, 2019, 201: 148-157. DOI:10.1016/j.atmosenv.2018.12.051 |
| [14] | Wang Z B, Wu Z J, Yue D L, et al. New particle formation in China:current knowledge and further directions[J]. Science of the Total Environment, 2017, 577: 258-266. DOI:10.1016/j.scitotenv.2016.10.177 |
| [15] | Gao J, Chai F H, Wang T, et al. Particle number size distribution and new particle formation:new characteristics during the special pollution control period in Beijing[J]. Journal of Environmental Sciences, 2012, 24(1): 14-21. |
| [16] | Quan J N, Liu Y G, Liu Q, et al. Anthropogenic pollution elevates the peak height of new particle formation from planetary boundary layer to lower free troposphere[J]. Geophysical Research Letters, 2017, 44(14): 7537-7543. DOI:10.1002/2017GL074553 |
| [17] | Huang X F, Wang C, Peng J F, et al. Characterization of particle number size distribution and new particle formation in Southern China[J]. Journal of Environmental Sciences, 2017, 51: 342-351. DOI:10.1016/j.jes.2016.05.039 |
| [18] | Huang X, Zhou L X, Ding A J, et al. Comprehensive modelling study on observed new particle formation at the SORPES station in Nanjing, China[J]. Atmospheric Chemistry and Physics, 2016, 16(4): 2477-2492. DOI:10.5194/acp-16-2477-2016 |
| [19] | Shen L J, Wang H L, Lü S, et al. Observation of aerosol size distribution and new particle formation at a coastal city in the Yangtze River Delta, China[J]. Science of the Total Environment, 2016, 565: 1175-1184. DOI:10.1016/j.scitotenv.2016.05.164 |
| [20] | Xiao S, Wang M Y, Yao L, et al. Strong atmospheric new particle formation in winter in urban Shanghai, China[J]. Atmospheric Chemistry and Physics, 2015, 15(4): 1769-1781. DOI:10.5194/acp-15-1769-2015 |
| [21] |
王红磊, 朱彬, 沈利娟, 等. 南京市夏季大气气溶胶新粒子生成事件分析[J]. 环境科学, 2012, 33(3): 701-710. Wang H L, Zhu B, Shen L J, et al. Atmospheric particle formation events in Nanjing during summer 2010[J]. Environmental Science, 2012, 33(3): 701-710. |
| [22] |
霍俊涛, 王新宁, 段玉森, 等. 2015~2017年上海郊区大气新粒子生成特征[J]. 环境科学, 2019, 40(11): 4791-4800. Huo J T, Wang X Y, Duan Y S, et al. First long-term study of atmospheric new particle formation in the suburb of Shanghai from 2015 to 2017[J]. Environmental Science, 2019, 40(11): 4791-4800. |
| [23] | Liu S, Hu M, Wu Z J, et al. Aerosol number size distribution and new particle formation at a rural/coastal site in Pearl River Delta (PRD) of China[J]. Atmospheric Environment, 2008, 42(25): 6275-6283. DOI:10.1016/j.atmosenv.2008.01.063 |
| [24] | Yue D L, Hu M, Wang Z B, et al. Comparison of particle number size distributions and new particle formation between the urban and rural sites in the PRD region, China[J]. Atmospheric Environment, 2013, 76: 181-188. DOI:10.1016/j.atmosenv.2012.11.018 |
| [25] |
岳玎利, 钟流举, 沈劲, 等. 珠三角地区大气新粒子增长-缩小过程特征[J]. 环境污染与防治, 2016, 38(3): 1-7. Yue D L, Zhong L J, Shen J, et al. Properties of new particle growth-shrinkage events in the Pearl River Delta region[J]. Environmental Pollution and Control, 2016, 38(3): 1-7. |
| [26] | Nie W, Ding A J, Wang T, et al. Polluted dust promotes new particle formation and growth[J]. Scientific Reports, 2015, 4: 6634. |
| [27] | Wang H L, Zhu B, Shen L J, et al. Number size distribution of aerosols at Mt. Huang and Nanjing in the Yangtze River Delta, China:effects of air masses and characteristics of new particle formation[J]. Atmospheric Research, 2014, 150: 42-56. DOI:10.1016/j.atmosres.2014.07.020 |
| [28] | Wu Z J, Hu M, Liu S, et al. New particle formation in Beijing, China:statistical analysis of a 1-year data set[J]. Journal of Geophysical Research:Atmospheres, 2007, 112(D9). DOI:10.1029/2006JD007406 |
| [29] | Zhu Y J, Sabaliauskas K, Liu X H, et al. Comparative analysis of new particle formation events in less and severely polluted urban atmosphere[J]. Atmospheric Environment, 2014, 98: 655-664. DOI:10.1016/j.atmosenv.2014.09.043 |
| [30] | Zhu Y J, Yan C Q, Zhang R Y, et al. Simultaneous measurements of new particle formation at 1 s time resolution at a street site and a rooftop site[J]. Atmospheric Chemistry and Physics, 2017, 17(15): 9469-9484. DOI:10.5194/acp-17-9469-2017 |
| [31] | Herrmann E, Ding A J, Kerminen V M, et al. Aerosols and nucleation in eastern China:first insights from the new SORPES-NJU station[J]. Atmospheric Chemistry and Physics, 2014, 14(4): 2169-2183. DOI:10.5194/acp-14-2169-2014 |
| [32] | Qi X M, Ding A J, Nie W, et al. Aerosol size distribution and new particle formation in the western Yangtze River Delta of China:2 years of measurements at the SORPES station[J]. Atmospheric Chemistry and Physics, 2015, 15(21): 12445-12464. DOI:10.5194/acp-15-12445-2015 |
| [33] | Wang D W, Guo H, Cheung K, et al. Observation of nucleation mode particle burst and new particle formation events at an urban site in Hong Kong[J]. Atmospheric Environment, 2014, 99: 196-205. DOI:10.1016/j.atmosenv.2014.09.074 |
| [34] | Lyu X P, Guo H, Cheng H R, et al. New particle formation and growth at a suburban site and a background site in Hong Kong[J]. Chemosphere, 2018, 193: 664-674. DOI:10.1016/j.chemosphere.2017.11.060 |
| [35] | Stein A F, Draxler R R, Rolph G D, et al. NOAA's HYSPLIT atmospheric transport and dispersion modeling system[J]. Bulletin of the American Meteorological Society, 2015, 96(12): 2059-2077. DOI:10.1175/BAMS-D-14-00110.1 |
| [36] | Kulmala M, Petäjä T, Nieminen T, et al. Measurement of the nucleation of atmospheric aerosol particles[J]. Nature Protocols, 2012, 7(9): 1651-1667. DOI:10.1038/nprot.2012.091 |
| [37] | Wang Z B, Hu M, Sun J Y, et al. Characteristics of regional new particle formation in urban and regional background environments in the North China Plain[J]. Atmospheric Chemistry and Physics, 2013, 13(24): 12495-12506. DOI:10.5194/acp-13-12495-2013 |
| [38] | Fuchs N A. The mechanics of aerosols[M]. New York: Dover Publications, 1989. |
| [39] | Gao J, Chai F H, Wang T, et al. Particle number size distribution and new particle formation (NPF) in Lanzhou, Western China[J]. Particuology, 2011, 9(6): 611-618. DOI:10.1016/j.partic.2011.06.008 |
| [40] | An J L, Wang H L, Shen L J, et al. Characteristics of new particle formation events in Nanjing, China:effect of water-soluble ions[J]. Atmospheric Environment, 2015, 108: 32-40. DOI:10.1016/j.atmosenv.2015.01.038 |
| [41] | Marti J J, Weber R J, McMurry P H, et al. New particle formation at a remote continental site:assessing the contributions of SO2 and organic precursors[J]. Journal of Geophysical Research:Atmospheres, 1997, 102(D5): 6331-6339. DOI:10.1029/96JD02545 |
| [42] | Chu B W, Zhang X, Liu Y C, et al. Synergetic formation of secondary inorganic and organic aerosol:effect of SO2 and NH3 on particle formation and growth[J]. Atmospheric Chemistry and Physics, 2016, 16(22): 14219-14230. DOI:10.5194/acp-16-14219-2016 |
| [43] | Carslaw D C, Rhys-Tyler G. New insights from comprehensive on-road measurements of NOx, NO2 and NH3 from vehicle emission remote sensing in London, UK[J]. Atmospheric Environment, 2013, 81: 339-347. DOI:10.1016/j.atmosenv.2013.09.026 |
| [44] | Carslaw D C. Evidence of an increasing NO2/NOx emissions ratio from road traffic emissions[J]. Atmospheric Environment, 2005, 39(26): 4793-4802. DOI:10.1016/j.atmosenv.2005.06.023 |
| [45] |
周凌晞, 汤洁, Ernst M, 等. 中国西部本底大气中CO的连续测量[J]. 环境科学, 2001, 22(3): 1-5. Zhou L X, Tang J, Ernst M, et al. Continuous measurement of baseline atmospheric carbon monoxide in western China[J]. Environmental Science, 2001, 22(3): 1-5. |
| [46] |
王丽涛, 张强, 郝吉明, 等. 中国大陆CO人为源排放清单[J]. 环境科学学报, 2005, 25(12): 1580-1585. Wang L T, Zhang Q, Hao J M, et al. Anthropogenic CO emission inventory of Mainland China[J]. Acta Scientiae Circumstantiae, 2005, 25(12): 1580-1585. |
| [47] |
田彪, 丁明虎, 孙维君, 等. 大气CO研究进展[J]. 地球科学进展, 2017, 32(1): 34-43. Tian B, Ding M H, Sun W J, et al. Research progress of atmospheric carbon monoxide[J]. Advances in Earth Science, 2017, 32(1): 34-43. |