 2020, Vol. 41
2020, Vol. 41

2. 华东理工大学资源与环境工程学院, 上海市环境保护化学污染物环境标准与风险管理重点实验室, 上海 200237;
3. 上海污染控制与生态安全研究院, 上海 200092
 , ZHANG Xue-wei1 , ZHOU Run-sheng1 , SONG Zhe1 , YAN Jia-ying1 , ZHOU Lei1,2,3
, ZHANG Xue-wei1 , ZHOU Run-sheng1 , SONG Zhe1 , YAN Jia-ying1 , ZHOU Lei1,2,3 , XIU Guang-li1,2,3
, XIU Guang-li1,2,3
2. Shanghai Key Laboratory of Environmental Standards and Risk Management of Chemical Pollutants, School of Resources and Environmental Engineering, East China University of Science and Technology, Shanghai 200237, China;
3. Shanghai Institute of Pollution Control and Ecological Security, Shanghai 200092, China
磺胺类抗生素(sulfonamides, SAs)是地表水中一种常见的污染物, 其被广泛应用于医疗、畜牧业和水产养殖业等[1]. SAs难以被动物体完全吸收, 绝大部分会通过粪尿排出体外从而进入环境[2], 导致其在世界范围内的检出率和检出浓度均相对较高[1].目前已有研究发现水环境中的SAs可能会通过诱导水生生物体内抗氧化酶活性的变化从而造成机体的氧化应激效应, 进而诱发水生生物的毒性[3].磺胺类抗生素在水体环境中的污染已成为一个日益严重的环境问题[4].
基于活化过硫酸盐(PS)和单过硫酸盐(PMS)产生硫酸根自由基(SO4- ·)的高级氧化技术(SR-AOP)是去除水中磺胺类抗生素的有效手段[5~7]. SR-AOP相对其他传统高级氧化技术因具有高效和安全稳定等优点在降解有机污染物方面得到了越来越多地研究应用[8].目前, 活化PS/PMS所需的手段包括了紫外光照射、加热、超声波活化和过渡金属活化等, 然而这些活化方式存在能耗大和易产生二次污染等问题, 限制了其在实际工程中的应用[9].例如, 在使用过渡金属活化过硫酸盐时, 过渡金属易形成沉淀同时造成二次污染[10]. SR-AOP作为高级氧化技术还可能会促成自由基与环境中其他物质的反应, 从而不可避免地形成副产物[9].因此, 为避免上述缺点, 利用基于单过硫酸盐非自由基机制降解污染物的方法开始受到人们的关注[11~13].
PMS具有1.82 V的标准氧化还原电位, 可以作为温和的氧化剂[14].利用其作为氧化剂的一大优势是PMS对特定的化合物表现出高度的选择性, 并能减少有毒有害副产物的产生[15].有研究发现PMS可以选择性直接氧化β-内酰胺类抗生素, 并且其对头孢氨苄(CFX)的降解效果甚至优于过渡金属活化PMS降解CFX的自由基方式[12].此外, Ji等[16]的研究发现非活化PMS能够直接氧化磺胺类抗生素磺胺甲唑(SMX), 主要反应活性位点为苯胺结构.这些研究都表明PMS作为直接氧化剂在处理抗生素废水方面具有相当广阔的前景.然而目前对于PMS直接氧化有机污染物的研究还不够充分[6, 12, 13, 17], 分析PMS非自由基的氧化过程仍具有重要的意义.
磺胺甲唑和柳氮磺胺吡啶(SSZ)同属于磺胺类抗生素, 但是分子结构有较明显的区别. SSZ已经在天然水体以及污水处理设施中大量检出, 其浓度范围大致在ng ·L-1~mg ·L-1之间[18].针对SSZ进行研究可以进一步补充PMS直接氧化磺胺类抗生素的相关动力学和机制.因此, 本研究选用SSZ作为目标有机物, 以非活化PMS为反应体系, 考察了PMS浓度、pH、离子强度、Cl-浓度和地表水对SSZ降解的影响, 同时对中间产物进行分析, 探讨了SSZ的降解机制, 以期为基于非活化PMS去除水中SSZ的应用可行性提供依据.
1 材料与方法 1.1 试剂柳氮磺胺吡啶(C18 H14 N4O5S, 98%)、过硫酸氢钾复合盐(Oxone, ≥47% KHSO5 basis)、磷酸二氢钾(KH2PO4)、碘化钠(NaI, ≥99.5%)均购自阿拉丁试剂有限公司;磷酸氢二钾(K2HPO4, 99%)购自上海迈瑞尔化学技术有限公司;磷酸钾(K3PO4, 98%)购自安耐吉化学;叔丁醇(C4H10 O, 99.5%)购自上海麦克林生化科技有限公司;硫代硫酸钠(Na2S2O3, 99%)购自九鼎化学;氢氧化钠(NaOH, ≥96%)、氯化钠(NaCl, ≥99.5%)购自GENERAL-REAGENT;2-2′-联氮双(3-乙基苯并噻唑啉-6-磺酸)二铵盐(ABTS, 99.91%)购自毕得药业, 5, 5-二甲基-1-吡咯啉-N-氧化物(DMPO, >98%)和2, 2, 6, 6-四甲基-4-哌啶醇(TEMP, >98%)购自Sigma-Aldrich.实验用水一般采用MQ超纯水(电阻率18.2 MΩ ·cm), 地表水样取自紫霞湖(32°06′ N, 118°84′ E).如无特殊说明, 实验中所用化学试剂均为分析纯, 未做进一步纯化处理.
1.2 实验方法所有反应均在100 mL玻璃瓶中进行, 反应温度为室温(20℃), 反应液中SSZ的初始浓度为20 μmol ·L-1, 溶液的pH值调节通过改变磷酸盐缓冲液的比例实现.反应进行时, 分别在0、30、90、150、210、270、300、330和360 min取出1 mL反应液和50 μL浓度为1 mol ·L-1的Na2S2O3混合以终止反应, 使用高效液相色谱仪(HPLC)测定溶液中SSZ浓度.在上述体系中分别考察PMS浓度(1、2、4、6和8 mmol ·L-1)、pH (5、6、7、8和9)、离子强度(10、30、40和50 mmol ·L-1)和地表水的影响.样品保存在4℃环境中并在24 h内完成HPLC分析.
1.3 SSZ和PMS的定量分析方法样品中SSZ浓度用Waters 2998型高效液相色谱仪进行分析.具体条件为:C18反相色谱柱(Agilent ZORBAX Eclipse Plus, 4.6×250 mm, 5 μm), 流动相为70% MeOH和30% H2 O (含0.1% H3PO4), 流速1 mL ·min-1, 柱温40℃, 进样量30 μL, 采用光电二极管阵列(PDA)检测器, 检测波长为345 nm.
PMS浓度用ABTS分光光度法测得[11, 19].将0.1 mL样品加入0.5 mL的ABTS (2 mmol ·L-1), 1 mL pH为4的醋酸缓冲液和20 μL浓度为1.5 mmol ·L-1的碘化钠的混合溶液中, 并用超纯水稀释至10 mL, 15 min后在UV-1700型紫外分光光度计中用415 nm波长测其吸光度.利用标准曲线换算得PMS浓度.
1.4 电子顺磁共振(EPR)检测本研究使用Bruker ELEXSYS E500型号的EPR搭配SHQ谐振器和Oxford ESR900连续流动低温恒温器对反应过程中产生的活性氧化物种进行检测.实验中使用DMPO作为羟基自由基(·OH)及硫酸根自由基(SO4- ·)的捕获剂, TEMP作为单线氧(1 O2)的捕获剂.将适量体积的样品分别加入DMPO和TEMP的水溶液中, 利用磷酸缓冲盐调节pH至7.将该溶液搅拌5 min后使用100 μL毛细管取样, 插入EPR腔中检测.检测条件为:20℃, 中心磁场磁通量密度为326 mT, 扫描宽度为10 mT, 扫描时间为30 s, 调制频率为100 kHz, 磁通量密度调制幅度为1 G(10-4T), 微波频率为9.63 GHz, 微波功率为10 mW.
1.5 中间产物鉴定配置50 mL反应液, 其中SSZ初始浓度为20 μmol ·L-1, PMS初始浓度为4 mmol ·L-1, pH为7.0.反应150 min后取出1 mL反应液加入50 μL浓度为1 mol ·L-1的Na2S2O3以终止反应.样品通过高分辨率质谱(HR-MS)分析, 使用正离子模式Waters Q-TOF质谱仪配合2998 PDA检测器检测, 色谱柱型号为KinetexTM C18, 100 mm~2.1 mm, 粒径2.6 μm, 孔径10 nm, 流速为0.2 mL ·min-1, 流动相为40% MeOH和60% H2 O (含0.1%甲酸).
2 结果与讨论 2.1 活性自由基鉴定预实验表明PMS能有效降解SSZ, 常温下(20℃)在SSZ初始浓度为20 μmol ·L-1的反应液(pH=7.0)中加入4 mmol ·L-1的PMS, 6 h后SSZ衰减至15.6 μmol ·L-1, 降解率为22%, 这表明在非活化条件下, PMS作为温和氧化剂能够在一定程度上去除SSZ.此外, PMS降解SSZ符合准一级反应动力学方程:
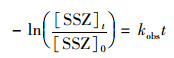
|
(1) |
式中, [SSZ]0为SSZ的初始浓度(μmol ·L-1), [SSZ]t为在反应时间t时, 反应体系中残余的SSZ浓度(μmol ·L-1), kobs为表观一级反应动力学常数(min-1), 其数值可以通过拟合实验数据获得.在上述条件下, 可以计算出SSZ的降解速率为1.78×10-3 min-1.
通常认为在PMS氧化体系中, ·OH及SO4- ·的生成会诱导有机污染物的降解[20, 21];此外, PMS在水相体系中, 还会发生降解反应生成1 O2[式(2)~(3)], 对污染物的降解存在潜在贡献[11, 22].
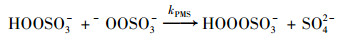
|
(2) |
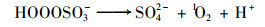
|
(3) |
本研究中, 为验证SO4- ·和·OH对SSZ降解的贡献程度, 在反应体系中引入了自由基淬灭剂叔丁醇(TBA)和甲醇(MeOH), 已知叔丁醇与SO4- ·和·OH的反应速率常数分别为(4~9.1)×105L ·(mol ·s)-1和(3.8~7.6)×108 L ·(mol ·s)-1, 甲醇与SO4- ·和·OH的反应速率常数分别为3.2×106L ·(mol ·s)-1和9.8×108 L ·(mol ·s)-1 [23].当在反应体系中分别加入过量(400 mmol ·L-1)叔丁醇和甲醇之后, SSZ的降解速率几乎未受影响.为了进一步验证SO4- ·与·OH的存在, 本研究还对反应体系进行了EPR检测, 如图 1 (b)所示, 在反应体系中未检测到SO4- ·与·OH的信号, 这表明未活化的PMS无法产生SO4- ·与·OH;相反, 利用TEMP捕获, 检测到了1 O2的信号[图 1 (a)].为了探究1 O2对SSZ降解的影响, 本研究对1 O2的贡献进行了估算.在pH=7.0时, 测得PMS自衰减速率为1.05×10-6 L ·(mol ·s)-1[19], 与文献[7]报道的数值一致.因此, 由PMS自降解产生1 O2的速率可以描述为:

|
反应条件:20℃, pH=7.0, [PMS]=4 mmol ·L-1, [SSZ]=20 μmol ·L-1, [缓冲盐]=10 mmol ·L-1 图 1 EPR检测非活化PMS体系中的活性氧化物种 Fig. 1 Detection of reactive oxygen species in unactivated PMS by EPR |
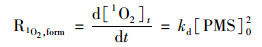
|
(4) |
式中, kd为PMS衰减速率常数, [PMS]0为PMS初始浓度(mmol ·L-1). PMS分解产生的1 O2, 除了能与目标污染物SSZ反应, 还能被水淬灭, 其中与SSZ反应的1 O2比例(f1O2, SSZ)等于:
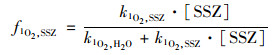
|
(5) |
式中, k1O2, SSZ是SSZ与1 O2的二级反应速率常数[1.47×107 L ·(mol ·s)-1], k1O2, H2O是水猝灭1 O2的速率常数(2.5×105 s-1)[11].因此反应初始阶段(0.5 h), 由1 O2诱导的SSZ降解速率可以描述为:
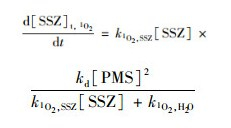
|
(6) |
计算结果表明反应体系中只有0.12%的1 O2与SSZ发生了反应, 由1 O2诱导的SSZ降解速率仅为2.0×10-14 L ·(mol ·s)-1, 远低于初始阶段SSZ的降解速率[2.0×10-10 L ·(mol ·s)-1].因此, 1 O2在非活化PMS体系中对SSZ降解的贡献可以忽略, 所以推断SSZ的衰减主要是由于PMS的直接氧化.
此外, 本研究还记录了反应过程中PMS的分解过程.在[PMS]0=4 mmol ·L-1, [SSZ]0=20 μmol ·L-1情况下, 未能检测到PMS浓度的显著降低, 主要原因是PMS浓度远大于SSZ, 即使SSZ全部反应, PMS的衰减量相对于其初始浓度而言几乎可以忽略.当使用浓度为1 mmol ·L-1的PMS和100 μmol ·L-1的SSZ进行反应时, 反应6 h后, PMS衰减了8%(图 2).

|
反应条件:20℃, pH=7.0, [PMS]=1 mmol ·L-1, [SSZ]= 100 μmol ·L-1, [缓冲盐]=10 mmol ·L-1 图 2 反应过程中PMS的衰减 Fig. 2 Degradation of PMS during reaction |
在SSZ初始浓度为20 μmol ·L-1, 反应温度为20℃, pH=7.0时, 将PMS的浓度从1 mmol ·L-1提升至8 mmol ·L-1会对SSZ的降解产生显著影响.如图 3 (a)所示, SSZ的降解速率随PMS浓度的升高而变快.本研究中, 1 mmol ·L-1 PMS条件下, 6 h内SSZ降解率仅为6%;当PMS剂量为8 mmol ·L-1时, 相同时间内SSZ的去除率超过40%.之前已有研究表明, 污染物的降解速率与PMS氧化剂的初始浓度成正比[16].在本研究的氧化体系中, PMS浓度远大于SSZ, PMS在反应过程中的消耗可忽略不计, 浓度基本维持不变. 图 3 (b)的拟合结果表明kobs与PMS浓度成正比, 表明该反应相对于PMS浓度是一级反应, 所以整体的反应符合二级动力学模型:

|
反应条件:20℃, pH=7.0, [SSZ]=20 μmol ·L-1, [缓冲盐]=10 mmol ·L-1 图 3 不同初始PMS浓度对SSZ降解的影响 Fig. 3 Effect of different PMS concentrations on the degradation of SSZ |
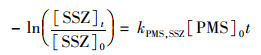
|
(7) |
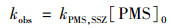
|
(8) |
式中, kPMS, SSZ为PMS与SSZ反应的二级反应速率常数L ·(mol ·h)-1, [PMS]0为PMS的初始浓度(mmol ·L-1), 在pH=7.0时, 可拟合出kPMS, SSZ数值为0.012 95 L ·(mol ·h)-1.
2.3 pH的影响如图 4所示, SSZ具有3个pKa值, 因此其在水溶液中具有4种不同的形态, 分别为分子态SSZ, 羧酸基团的去质子化形态SSZ-H-(pKa1=3.108), 磺酰胺基团的去质子化形态SSZ-2H2-(pKa2=6.505)和酚基基团的去质子化形态SSZ-3H3-(pKa3=9.371)[25].每种形态的浓度分数取决于pH, 可以通过下式描述:

|
图 4 SSZ、SSZ-H-、SSZ-2H2-和SSZ-3H3-的结构式 Fig. 4 Molecular structures of SSZ, SSZ-H-, SSZ-2H2-, and SSZ-3H3- |
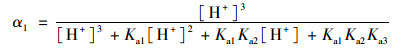
|
(9) |
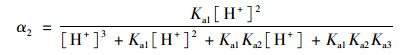
|
(10) |
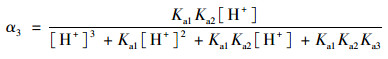
|
(11) |
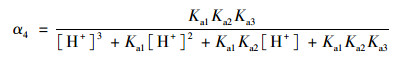
|
(12) |
式中, α1、α2、α3和α4分别是SSZ、SSZ-H-、SSZ-2H2-和SSZ-3H3-的摩尔分数. SSZ的总浓度[SSZ]tot可以用以下公式计算:
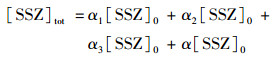
|
(13) |
PMS具有两个pKa, 其中pKa1 < 0, pKa2=9.4[26].因此, 在所测pH范围内(5~9), PMS主要以HSO5-和SO52-形式存在. PMS的总浓度[PMS]tot可以用式(14)计算:
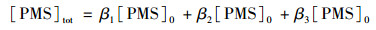
|
(14) |
式中, β1、β2、和β3, 分别是H2SO5、HSO5-和SO52-的摩尔分数. SSZ的转化速率可用下式计算:
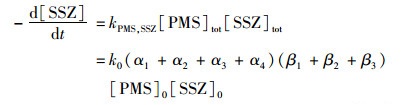
|
(15) |
由于在研究的pH范围内α1、β1过小可忽略不计, 因此kPMS, SSZ可简化为:

|
(16) |
因此, PMS与SSZ反应的二阶速率常数kPMS, SSZ可简化为:
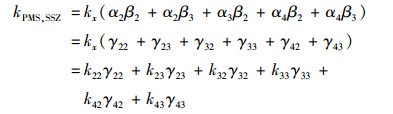
|
(17) |
式中, γ22、γ23、γ32、γ33、γ42和γ43分别是SSZ-H-与HSO5-、SSZ-H-与SO52-、SSZ-2H2-与HSO5-、SSZ-2H2-与SO52-、SSZ-3H3-与HSO5-和SSZ-3H3-与SO52-之间的反应在总反应中的摩尔分数.由于γ23和γ42接近0可忽略不计, γ43在pH小于8时接近0, γ22在pH大于8时接近0, 所以5 < pH≤8时式(17)可写为:
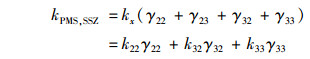
|
(18) |
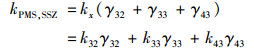
|
(19) |
式中, k22、k32、k33和k43的值通过式(19)和动力学数据的最小二乘非线性回归计算得到.动力学模型计算得到的k22、k32、k33和k43分别为0.056 1、0.094 9、0.150和0.281 L ·(mol ·s)-1.所以, 在pH 5~8时, SSZ-2H2-与SO52-之间的反应占主导, 在pH 8~9范围内, SSZ-3H3-与SO52-之间的反应占主导.因此, SSZ的SSZ-3H3-形态与SO52-之间的氧化更具反应性. pH的升高导致了SSZ-3H3-和SO52-在溶液中比例的增加, 从而有助于PMS降解SSZ.
一般认为溶液的酸碱度对氧化反应起重要作用, pH值能够影响氧化剂和目标污染物的赋存形态及浓度, 进而影响氧化反应过程[25]. 图 5表明, 在SSZ初始浓度为20 μmol ·L-1, PMS初始浓度为4 mmol ·L-1, 反应温度为20℃时, 随着溶液pH值的升高, SSZ的降解效率提高.相比于酸性条件, 碱性条件更有利于SSZ的降解, 这与文献[13]中报道的PMS直接氧化氟喹诺酮类抗生素所得结论一致.

|
反应条件:20℃, [PMS]=4 mmol ·L-1, [SSZ]=20 μmol ·L-1, [缓冲盐]=10 mmol ·L-1 图 5 不同初始pH对SSZ降解的影响 Fig. 5 Effect of different initial pH on the degradation of SSZ |
离子强度的变化会影响PMS直接氧化的反应速率[11].本研究通过改变磷酸缓冲盐的浓度来改变反应体系的离子强度, 进而探究不同离子强度对SSZ降解的影响.如图 6所示, 在20℃、pH=7.0, [PMS]=4 mmol ·L-1、[SSZ]=20 μmol ·L-1条件下, 随着缓冲盐浓度从10 mmol ·L-1提高到50 mmol ·L-1,kobs也随之升高. Lou等[27]的研究发现磷酸盐会活化PMS产生羟基自由基从而促进污染物的去除, 另外也有研究发现磷酸盐能够促进PMS对AO7的脱色效果[28].目前关于离子强度对SSZ降解的促进机制尚不明确, 有待进一步研究.

|
反应条件:20℃, pH=7.0, [PMS]=4 mmol ·L-1, [SSZ]=20 μmol ·L-1 图 6 离子强度对SSZ降解的影响 Fig. 6 Effect of ionic strength on the degradation of SSZ |
Cl-是天然水体的组成部分[29], 同时也广泛存在于各种工业废水中[30~32], 因此有必要讨论Cl-对PMS直接氧化SSZ的影响.本研究结果表明, Cl-的存在显著提高了SSZ的降解效率;同时, 提高氯离子浓度有利于SSZ的去除(图 7). Lei等[33]在研究中发现, Cl-在水相中会和PMS发生一系列[式(20)~(23)]反应, 生成氧化性较强的- OCl从而促进了SSZ的降解.

|
反应条件: 20℃, pH=7.0, [PMS]=4 mmol ·L-1, [SSZ]=20 μmol ·L-1, [缓冲盐]=10 mmol ·L-1 图 7 不同的Cl-浓度对SSZ降解的影响 Fig. 7 Effect of different Cl- On the degradation of SSZ |

|
(20) |

|
(21) |
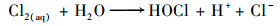
|
(22) |
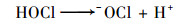
|
(23) |
值得注意的是, Cl-的存在虽然促进了SSZ的降解, 但同时也增加了这类磺胺类抗生素降解产物的毒性, 如生成各类氯代消毒副产物等[34].因此在非活化PMS降解有机污染物的实际应用中还应考虑其氯化产物的进一步处理.
2.6 地表水影响地表水组分复杂[35], 会对非活化PMS的实际应用产生影响.因此, 为探究非活化PMS体系在实际应用中的处理效果, 本研究在地表水样中对SSZ进行了降解实验.本实验中采用的地表水样为紫霞湖水样, 其水质参数如表 1所示.在反应前使用NaOH溶液调节反应体系的pH至7.如图 8所示, 在反应6 h后, 控制组中SSZ降解率为37%, 地表水组中SSZ降解率为22%, 这表明地表水会明显抑制PMS对SSZ的降解. Ji等[16]在使用非活化PMS降解磺胺类抗生素的研究中同样发现, 地表水会对非活化PMS体系产生抑制.推测在本体系中产生抑制的可能原因是紫霞湖水样中的TOC较高.

|
反应条件:20℃, pH=7.0, [PMS]=4 mmol ·L-1, [SSZ]=20 μmol ·L-1 图 8 地表水对PMS直接降解SSZ的影响 Fig. 8 Effect of surface water on direct degradation of SSZ by PMS |
|
|
表 1 湖水水质成分 Table 1 Characteristic of lake water |
2.7 反应位点鉴定
为了进一步探究SSZ与PMS反应的机制, 本研究还引入了SSZ的结构相似物SPD及5-ASA(结构如图 9所示), 分别探究他们与PMS的反应活性.本实验结果表明, PMS对5-ASA和SPD的氧化活性远远高于SSZ. Ji等[16]在研究PMS直接氧化磺胺二甲吡啶和磺胺甲唑中发现, 反应的主要活性位点是苯胺基团. SPD及5-ASA都含有苯胺基团, 因此可以解释为何SPD及5-ASA与PMS直接反应的活性相对较高, 而含有偶氮基团的SSZ则相对惰性.前沿电子轨道计算结果表明SSZ的7N, 19C和25O位置(图 4)电子云密度相对较高, 易受氧化剂的亲电攻击[31], 因此推断这3个位置是PMS直接氧化SSZ的反应活性位点.

|
图 9 PMS降解SPD、SSZ和5-ASA二级反应速率常数的对比 Fig. 9 Comparison of the second order rate constant of SPD, SSZ, and 5-ASA by PMS |
通过高分辨率质谱(HR-MS)分析非活化PMS降解SSZ的反应样品以鉴定反应产物以及反应途径, 产物鉴定结果如表 2所示.质子化SSZ在m/z=399.076 7时表现出离子峰;产物2的相对分子质量和母体相比损失了63.960 8, 结合相对分子质量与分子式可以推断为SSZ中SO2基团被挤脱之后的产物(SSZ-SO2);产物4相对于母体增加了15.993 7, 可以推断为羟基化产物(SSZ+OH);而产物3(m/z=351.110 6)则可以理解为SO2挤脱与羟基化同时发生之后生成的产物(SSZ-SO2+OH).结合以上产物分析, 推测非活化PMS直接氧化SSZ的途径主要有2条, 如图 10所示.第一条途径是羟基化过程, PMS亲电攻击SSZ右侧苯环上的19C或25O, 与羟基相邻的苯环和溶液中的OH-发生亲电取代反应生成加羟基产物SSZ+OH;第二条降解途径为SO2的挤脱反应, 也称作Smiles型重排, 这种分子结构内的重排作用发生于与磺酰胺基团相连的苯环和吡啶环之间, 伴随着S—C键断裂和新的N—C键的生成[6, 18, 34].此过程在含六元杂环磺胺类药物分子的氧化过程中较为常见, 在高铁酸盐, 硫酸根自由基, 羟基自由基氧化该类物质中均有报道[6, 36, 37].
|
|
表 2 SSZ降解产物的质谱数据和推测结构 Table 2 Products detected during the degradation of SSZ |

|
图 10 非活化PMS降解SSZ的反应途径 Fig. 10 Proposed transformation pathways for SSZ degradation by non-activated PMS |
非活化PMS可以降解水溶液中的磺胺类抗生素SSZ, 降解过程符合准一级反应动力学规律.增加PMS浓度, 可有效提高SSZ的降解速率, 且表观反应速率常数与PMS的浓度成正比. SSZ的降解还受到溶液pH、离子强度和氯离子的影响.本研究表明, 碱性条件有利于反应的进行;提高反应体系的离子强度也能够促进SSZ的降解. Cl-的存在对SSZ的降解有显著的提高, 低浓度的Cl-就能大幅度促进反应的进行;地表水会抑制SSZ的降解. SSZ的7N、19C和25O这3个位置电子云密度相对较高, 易受PMS的亲电攻击. SSZ的主要降解途径包括了羟基化和SO2挤脱.
| [1] | Kümmerer K. Antibiotics in the aquatic environment-A review-Part I[J]. Chemosphere, 2009, 75(4): 417-434. |
| [2] |
陈姗, 许凡, 张玮, 等. 磺胺类抗生素污染现状及其环境行为的研究进展[J]. 环境化学, 2019, 38(7): 1557-1569. Chen S, Xu F, Zhang W, et al. Research progress in pollution situation and environmental behavior of sulfonamides[J]. Environmental Chemistry, 2019, 38(7): 1557-1569. |
| [3] | Wang N, Noemie N, Nguyen-Ngoc H, et al. Adverse effects of enrofloxacin when associated with environmental stress in Tra catfish (Pangasianodon hypophthalmus)[J]. Chemosphere, 2009, 77(11): 1577-1584. |
| [4] |
徐颖, 王祺, 苏墨, 等. 磺胺抗生素在河流沉积物中的吸附特性研究[J]. 安全与环境学报, 2016, 16(2): 278-283. Xu Y, Wang Q, Su M, et al. Adsorption characteristics of sulfa antibiotics in the river sediments[J]. Journal of Safety and Environment, 2016, 16(2): 278-283. |
| [5] |
周景尧, 李哲, 陈家玮. 共存重金属离子对针铁矿活化过硫酸盐去除水中磺胺吡啶的影响[J]. 地学前缘, 2019, 26(4): 295-300. Zhou J Y, Li Z, Chen J W. Effect of coexisting heavy metal ions on the activation of persulfate by goethite for the removal of sulfapyridine from aqueous solutions[J]. Earth Science Frontiers, 2019, 26(4): 295-300. |
| [6] | Jiang M D, Zhang Q Y, Ji Y F, et al. Transformation of antimicrobial agent sulfamethazine by peroxymonosulfate:Radical vs. nonradical mechanisms[J]. Science of the Total Environment, 2018, 636: 864-871. |
| [7] | Lou X Y, Fang C L, Geng Z N, et al. Significantly enhanced base activation of peroxymonosulfate by polyphosphates:kinetics and mechanism[J]. Chemosphere, 2017, 173: 529-534. |
| [8] | Wang J L, Wang S Z. Activation of persulfate (PS) and peroxymonosulfate (PMS) and application for the degradation of emerging contaminants[J]. Chemical Engineering Journal, 2018, 334: 1502-1517. |
| [9] |
许芬, 陈家斌, 张书源, 等. 丙酮活化过一硫酸盐性能及非自由基机制[J]. 环境科学学报, 2018, 38(11): 4333-4339. Xu F, Chen J B, Zhang S Y, et al. Performance and non-radical mechanism of activation of peroxymonosulfate by acetone[J]. Acta Scientiae Circumstantiae, 2018, 38(11): 4333-4339. |
| [10] |
蒋梦迪, 张清越, 季跃飞, 等. 热活化过硫酸盐降解三氯生[J]. 环境科学, 2018, 39(4): 1661-1667. Jiang M D, Zhang Q Y, Ji Y F, et al. Degradation of triclosan by heat activated persulfate oxidation[J]. Environmental Science, 2018, 39(4): 1661-1667. |
| [11] | Yang Y, Banerjee G, Brudvig G W, et al. Oxidation of organic compounds in water by unactivated peroxymonosulfate[J]. Environmental Science & Technology, 2018, 52(10): 5911-5919. |
| [12] | Chen J B, Fang C, Xia W J, et al. Selective transformation of β-Lactam antibiotics by peroxymonosulfate:reaction kinetics and nonradical mechanism[J]. Environmental Science & Technology, 2018, 52(3): 1461-1470. |
| [13] | Zhou Y, Gao Y, Pang S Y, et al. Oxidation of fluoroquinolone antibiotics by peroxymonosulfate without activation:kinetics, products, and antibacterial deactivation[J]. Water Research, 2018, 145: 210-219. |
| [14] | Betterton E A, Hoffmann M R. Kinetics and mechanism of the oxidation of aqueous hydrogen sulfide by peroxymonosulfate[J]. Environmental Science & Technology, 1990, 24(12): 1819-1824. |
| [15] | Zhang T, Chen Y, Wang Y R, et al. Efficient peroxydisulfate activation process not relying on sulfate radical generation for water pollutant degradation[J]. Environmental Science & Technology, 2014, 48(10): 5868-5875. |
| [16] | Ji Y F, Lu J H, Wang L, et al. Non-activated peroxymonosulfate oxidation of sulfonamide antibiotics in water:kinetics, mechanisms, and implications for water treatment[J]. Water Research, 2018, 147: 82-90. |
| [17] | Yin R L, Guo W Q, Wang H Z, et al. Selective degradation of sulfonamide antibiotics by peroxymonosulfate alone:direct oxidation and nonradical mechanisms[J]. Chemical Engineering Journal, 2018, 334: 2539-2546. |
| [18] | Ji Y F, Yang Y, Zhou L, et al. Photodegradation of sulfasalazine and its human metabolites in water by UV and UV/peroxydisulfate processes[J]. Water Research, 2018, 133: 299-309. |
| [19] | Yang Y, Jiang J, Lu X L, et al. Production of sulfate radical and hydroxyl radical by reaction of ozone with peroxymonosulfate:a novel advanced oxidation process[J]. Environmental Science & Technology, 2015, 49(12): 7330-7339. |
| [20] | Anipsitakis G P, Dionysiou D D. Radical generation by the interaction of transition metals with common oxidants[J]. Environmental Science & Technology, 2004, 38(13): 3705-3712. |
| [21] | Zhou Y, Jiang J, Gao Y, et al. Activation of peroxymonosulfate by benzoquinone:a novel nonradical oxidation process[J]. Environmental Science & Technology, 2015, 49(21): 12941-12950. |
| [22] | Ball D L, Edwards J O. The kinetics and mechanism of the decomposition of caro's acid[J]. Journal of the American Chemical Society, 1956, 78(6): 1125-1129. |
| [23] | Liang C J, Su H W. Identification of sulfate and hydroxyl radicals in thermally activated persulfate[J]. Industrial & Engineering Chemistry Research, 2009, 48(11): 5558-5562. |
| [24] | Yılmaz H, Üstün Z, Demiralay E C. RPLC determination of acid dissociation constants and quantitative estimation for sulfasalazine[J]. Journal of the Iranian Chemical Society, 2016, 13(1): 103-110. |
| [25] | Sharma V K, Mishra S K, Nesnas N. Oxidation of sulfonamide antimicrobials by ferrate(VI)[FeVIO42-[J]. Environmental Science & Technology, 2006, 40(23): 7222-7227. |
| [26] | Guan Y H, Ma J, Li X C, et al. Influence of pH on the formation of sulfate and hydroxyl radicals in the UV/Peroxymonosulfate system[J]. Environmental Science & Technology, 2011, 45(21): 9308-9314. |
| [27] | Lou X Y, Wu L X, Guo Y G, et al. Peroxymonosulfate activation by phosphate anion for organics degradation in water[J]. Chemosphere, 2014, 117: 582-585. |
| [28] | Qi C D, Liu X T, Ma J, et al. Activation of peroxymonosulfate by base:implications for the degradation of organic pollutants[J]. Chemosphere, 2016, 151: 280-288. |
| [29] |
彭安, 王子健, 许坤. 萃取法测定氯化汞络合物的稳定性常数[J]. 环境科学, 1982, 3(4): 9-13. Peng A, Wang Z J, Xu K. Determination of stability constants of mercuric chloride complex by extraction method[J]. Environmental Science, 1982, 3(4): 9-13. |
| [30] |
张春磊, 杨志成, 李丽婷. 高氯废水中氯离子的去除研究[J]. 化工管理, 2018(20): 26. Zhang C L, Yang Z C, Li L T. Study on dechlorination of high chlorine wastewater[J]. Chemical Enterprise Management, 2018(20): 26. |
| [31] |
唐宝玲, 左松, 陈胜文, 等. 石灰铝盐法沉淀废水中高浓度氯离子的研究[J]. 上海第二工业大学学报, 2018, 35(4): 255-262. Tang B L, Zuo S, Chen S W, et al. Study on precipitation of high concentration chloride ion in wastewater by lime with aluminum salt[J]. Journal of Shanghai Polytechnic University, 2018, 35(4): 255-262. |
| [32] |
郭亚丽, 李红. 硝酸银容量法测定浆液中氯离子含量探讨[J]. 全面腐蚀控制, 2018, 32(4): 4-6. Guo Y L, Li H. Investigation of determination chloride ions in FGD waste water with silver nitrate volumetric[J]. Total Corrosion Control, 2018, 32(4): 4-6. |
| [33] | Lei Y, Chen C S, Ai J, et al. Selective decolorization of cationic dyes by peroxymonosulfate:non-radical mechanism and effect of chloride[J]. RSC Advances, 2016, 6(2): 866-871. |
| [34] | Fu W J, Li B, Yang J Q, et al. New insights into the chlorination of sulfonamide:smiles-type rearrangement, desulfation, and product toxicity[J]. Chemical Engineering Journal, 2018, 331: 785-793. |
| [35] |
唐晓璐. 水污染现状及其处理的新技术[J]. 中小企业管理与科技, 2017(25): 193-196. Tang X L. The present situation of water pollution and the new treatment technology[J]. Management & Technology of SME, 2017(25): 193-196. |
| [36] | Fan X Q, Hao H Y, Shen X X, et al. Removal and degradation pathway study of sulfasalazine with Fenton-like reaction[J]. Journal of Hazardous Materials, 2011, 190(1-3): 493-500. |
| [37] | Feng M B, Baum J C, Nesnas N, et al. Oxidation of sulfonamide antibiotics of six-membered heterocyclic moiety by ferrate(VI):Kinetics and mechanistic insight into SO2 extrusion[J]. Environmental Science & Technology, 2019, 53(5): 2695-2704. |