 2020, Vol. 41
2020, Vol. 41



2. 昆山市建筑设计有限公司, 昆山 215300;
3. 同济大学环境科学与工程学院, 上海 200092
2. Kunshan Architectural Design Co., Ltd., Kunshan 215300, China;
3. College of Environmental Science and Engineering, Tongji University, Shanghai 200092, China
人工合成染料广泛应用于纺织、皮革制造以及造纸等领域.偶氮染料因其分子结构中通常含有偶氮键(—N=N—)而得名.世界染料总产量每年超过1万t, 其中15%以上在染色过程中损失[1], 由于其难以生物降解和潜在的致癌性, 对环境造成了严重的污染, 是严重危害人体健康的因素[2].而常见的处理工艺, 如吸附和絮凝, 并不能有效去除偶氮染料带来的污染[3].
近年来, 一些基于H2O2产生羟基自由基(·OH)的新型高级氧化技术(AOPs)被开发用于降解饮用水和工业废水中的难生物降解有机物[4, 5].然而H2O2不稳定的特性限制了其大规模的应用.固体状态的过碳酸钠(SPC)(Na2CO3·1.5H2O2)被认为比H2O2更加稳定[6], 可作为H2O2的“干载体”, 具有在室温下操作简单、安全稳定的特性.同时, SPC拥有比H2O2更宽的pH适应能力以及更高的污染物去除效率[7].使用Fe2+、Fe3+和V4+[8, 9]等金属离子, 以及使用螯合剂强化Fe3+[10]活化SPC用于高级氧化的均相体系已经见诸报道, 但这些体系依旧存在铁盐沉淀和有害金属离子渗漏进入环境的问题.为了解决这些问题, 使用Fe单质[11]、Fe3O4[12]和沸石负载Fe/Cu双金属[13]活化SPC的非均相体系逐渐受到关注.但这些基于Fe、Cu及其氧化物的非均相体系存在着催化效率较低或回收较为困难的不足.在高级氧化技术中CuO展现了良好的催化活性[14], 为了防止其Cu离子的渗出, 降低其回收难度, 其中一种可能的方法是将CuO与Fe3O4结合, 同时利用了CuO的高催化性能和Fe3O4的磁性并且有可能产生双金属氧化物的协同效应, 进一步提高Fe3O4-CuO复合材料的催化能力.
本文采用合成的Fe3O4-CuO材料催化SPC体系降解偶氮染料AO7.考察了Fe3O4-CuO投加量, SPC投加量, 溶液初始pH以及染料废水中常见的Cl-离子和天然有机质(NOM)对反应体系的影响.通过猝灭实验研究反应机制, 并进行了材料重复利用实验, 考察了Fe3O4-CuO在实际应用中的潜力.
1 材料与方法 1.1 实验材料金橙Ⅱ(AO7)购于国药集团化学试剂有限公司(相关参数见表 1), 过碳酸钠(SPC)(Na2CO3·1.5H2O2)购于Alfa Aesar化学有限公司;硫酸(H2SO4)、氢氧化钠(NaOH)、硫酸亚铁(FeSO4·7H2O)、硫酸铜(CuSO4·5H2O)、亚硝酸钠(NaNO2), 氯化钠(NaCl)、叔丁醇(C4H9OH)、苯酚(C6H5OH)和腐殖酸(HA)均为分析纯;硝酸(HNO3)为优级纯.
|
|
表 1 染料的相关参数 Table 1 Elated parameters of dyes |

1.2 实验方法 1.2.1 Fe3O4-CuO材料的合成
Fe3O4-CuO催化剂采用简单的一步水热法制备.制备过程可以简单地表示如下:烧杯置于恒温水浴锅中保持80℃, 在连续通入空气吹扫(流速=3.0 L·min-1)的条件下, 将CuSO4·5H2O(500 mL, 0.1 mol·L-1)和FeSO4·7H2O(500 mL, 0.5 mol·L-1)混合.在反应过程中使用0.1 mol·L-1的H2SO4和NaOH调节溶液pH使其保持pH=8.0.反应结束后所需固体用磁铁分离, 每次用1.5 L去离子水洗涤3次, 并在105℃下干燥[15].单独的Fe3O4和CuO同样按照该反应条件合成.
1.2.2 AO7降解实验AO7在不同体系下的降解:反应液的体积为100 mL, AO7的初始浓度为0.05 mmol·L-1, 加入一定量的Fe3O4-CuO材料, 使用0.01 mol·L-1的H2SO4和NaOH调节pH到设定值后再加入SPC启动反应.在预定时间取样并使用0.20 mol·L-1的NaNO2猝灭终止反应并测定剩余染料的吸光度.整个反应置于25℃的恒温水浴摇床中进行.
1.3 分析方法AO7浓度采用Mapada UV 1600(PC)紫外可见光分光光度计, 于AO7最大吸收波长484 nm处测定经过0.45 μm滤头过滤的滤液吸光度, 代入标准曲线计算对应浓度c.采用WTW inLab pH7110型pH计测定pH;采用岛津AA 6300火焰原子吸收光谱仪(AAS)测定反应液中游离金属离子浓度;采用岛津TOC-L总有机碳分析仪(TOC)测定降解过程中总有机碳的变化;使用Hitachi S4800扫描电子显微镜联用能谱仪(SEM-EDS)和Rigaku TTRAX Ⅲ型X射线粉末衍射仪(XRD)对Fe3O4-CuO磁性材料进行表征.使用JEOL FA200型电子自旋共振顺磁波谱仪(EPR)对反应过程中产生的自由基进行鉴定.
2 结果与讨论 2.1 Fe3O4-CuO磁性材料的SEM-EDS分析由图 1可以看出Fe3O4-CuO颗粒呈现球状结构, 表面粗糙孔隙较多并且有明显的团聚现象.表 2的EDS结果显示了材料中不同元素的占比, 清晰地表明成功合成出Fe3O4-CuO磁性材料.

|
(a) ×25 000; (b) × 100 000, 图 1 Fe3O4-CuO材料的SEM图 Fig. 1 SEM image of Fe3O4-CuO |
|
|
表 2 Fe3O4-CuO中不同元素的含量 Table 2 Content of different elements in Fe3O4-CuO |
2.2 Fe3O4-CuO磁性材料的XRD分析
Fe3O4-CuO磁性材料的XRD衍射图谱如图 2所示.从中可以看出, 合成材料的衍射峰与磁铁矿型Fe3O4(JCPDS19-0629)匹配度良好, 2θ角分别在18.3°、30.1°、35.4°、43.4°、53.1°、56.9°、62.5°和75.0°处的衍射峰分别对应了磁铁矿型Fe3O4的(111)、(220)、(311)、(400)、(422)、(511)、(440)和(622)面的衍射峰.在38.5°、42.2°、61.3°和72.2°处的衍射峰和氧化铜型CuO(JCPDS44-0746)匹配良好.

|
图 2 Fe3O4-CuO材料的XRD图谱 Fig. 2 XRD patterns of Fe3O4-CuO |
对3种体系降解AO7的能力进行对比实验, 结果如图 3.当体系中仅投加Fe3O4-CuO磁性材料时, 在30 min的总反应时长内, AO7的去除率仅为11%, 这主要是因为Fe3O4-CuO材料可以对AO7产生一定的吸附效果.当体系中投加2.0 mmol·L-1的SPC时, 相同时间内反应体系中AO7浓度未发生变化, 说明单独SPC并不能有效降解AO7.当同时投加0.2 g·L-1 Fe3O4-CuO和2.0 mmol·L-1 SPC时, 在20 min内去除率已经达到100%, 两者显示出了明显的协同效果, 推测是因为Fe3O4-CuO材料可以有效活化SPC溶于水后产生的H2O2, 产生自由基对AO7进行降解.

|
[A07]0=0.10 mmol·L-1; pH0=7.0 图 3 不同体系中AO7的降解效果 Fig. 3 Degradation effect of AO7 under different systems |
保持SPC与AO7的量比为20:1, 改变Fe3O4-CuO材料投加量以研究材料浓度对AO7降解实验的影响, 实验结果如图 4, 各组一级反应速率常数见表 3.

|
[AO7]0=0.10 mmol·L-1; [SPC]0=2.0 mmol·L-1; pH0=7.0 图 4 不同材料投加量对AO7的降解效果 Fig. 4 Effect of Fe3O4-CuO loading on AO7 degradation |
|
|
表 3 不同Fe3O4-CuO投加量的一级反应速率常数 Table 3 Reaction rate constants at different Fe3O4-CuO loadings |
由图 4可知, Fe3O4-CuO材料在反应体系中起到活化SPC降解AO7的作用.由表 3可知, 反应的一级反应速率常数与材料投加量的升高呈线性关系.随着材料投加量的上升, AO7的降解速度明显加快.这主要是因为Fe3O4-CuO材料对SPC产生了活化效果, 随着材料投加量的上升, 更多的材料提供了更多的反应位点, SPC被活化的速度加快从而提高了AO7的降解速度.
2.5 SPC投加量的影响保持Fe3O4-CuO材料投加量为0.2 g·L-1, 逐步增加SPC投加量, 以研究氧化剂投加量对AO7降解的影响, 实验结果如图 5, 各组一级反应速率常数见表 4.

|
[AO7]0=0.10 mmol·L-1; [Fe3O4-CuO]0=0.2 g·L-1; pH0=7.0 图 5 不同氧化剂投加量对AO7的降解效果 Fig. 5 Degradation effect of AO7 under different amounts of oxidant |
|
|
表 4 不同SPC投加量的一级反应速率常数 Table 4 Reaction rate constant under different SPC concentration |
由表 4可知当SPC投加量由1.0 mmol·L-1SPC上升到3.0 mmol·L-1时, 反应速率常数由0.064 1 min-1上升到了0.213 3 min-1, 这是因为当SPC溶于水后会产生大量的H2O2 [式(1)], 随着投加量的上升, 由Fe3O4-CuO材料活化H2O2产生的自由基增多, 加快了反应的进行;同时随着SPC投加量的上升, 当SPC完全溶于水后反应体系的pH值会发生变化, 由SPC浓度1.0 mmol·L-1时的pH=9.62上升到3.0 mmol·L-1的pH=9.92.反应体系中pH的增大同样会加快H2O2被活化的速度, 具体过程见2.6节反应体系中pH影响讨论.
当SPC投加量上升至4.0 mmol·L-1时, 反应速率增加的速度开始减缓, 仅上升到0.228 3 min-1;投加量继续上升后反应反而受到抑制, 当投加量为5 mmol·L-1反应速率下降到0.123 8 min-1, 继续增大至6.0 mmol·L-1时反应速率下降到0.100 0 min-1.这是因为当溶液中过量的H2O2被材料活化产生·OH, 此时反应体系中过量的·OH会发生自我猝灭[式(2)], 同时会与H2O2反应生成O2-·[式(3)], 产生的O2-·进一步消耗·OH反应生成1O2 [式(4)].这些反应均会消耗·OH, 对AO7的降解产生抑制作用[16~18].

|
(1) |

|
(2) |

|
(3) |

|
(4) |
控制AO7、Fe3O4-CuO和SPC的初始浓度, 改变反应初始pH值, 以研究pH对反应的影响, 实验结果如图 6.在反应中测量溶液pH值的变化情况, 结果如图 7.采用质量滴定法测量材料的零电荷点(pHPZC)[19], 结果如图 8.

|
[AO7]0=0.10 mmol·L-1; [Fe3O4-CuO]0=0.2 g·L-1; [SPC]0=2.0 mmol·L-1 图 6 不同初始pH对AO7降解的影响 Fig. 6 Effects of initial pH on AO7 degradation |

|
[AO7]0=0.10 mmol·L-1; [Fe3O4-CuO]0=0.2 g·L-1; [SPC]0=2.0 mmol·L-1 图 7 反应中pH值的变化情况 Fig. 7 Variation of pH values |

|
图 8 Fe3O4-CuO材料pHPZC的测量 Fig. 8 pHPZC of Fe3O4-CuO |
由图 6可知, 随着初始pH的增大, AO7的脱色速度逐渐加快, pH=9.0时效果最好.由图 7可知, 由于SPC具有较强的缓冲能力, 无论初始pH值大小, 均能快速在反应液中形成碱性环境, 且pH值均大于Fe3O4-CuO的零电荷点6.02.当溶液pH>pHPZC时, 材料表面带负电荷, 而AO7为阴离子染料, 所以物质间的静电作用力不是造成不同初始pH条件下降解速度不同的主要原因.根据研究表明, 在碱性条件下, H2O2会被电离形成氢过氧化物阴离子HO2- [式(5)], 并被分解产生活性氧物种, 如羟基自由基·OH [式(6)]、超氧自由基O2-·[式(7)]和单线态氧1O2[式(8)][20, 21].SPC溶于水所提供的碱性环境加快了自由基的产生, 有利于AO7更加快速地降解.

|
(5) |

|
(6) |

|
(7) |

|
(8) |
为考察Fe3O4-CuO/SPC体系降解AO7的反应机制, 进行自由基猝灭实验, 结果如图 9.SPC溶于水后产生H2O2, 一般认为会被活化产生·OH.叔丁醇(TBA)是·OH的优良猝灭剂, TBA与·OH反应速率可达(3.8~7.6)×108 mol-1·s-1[22], 常用于鉴定反应体系中是否产生了·OH.当投加0、50和500 mmol·L-1叔丁醇时, 反应30 min后AO7去除率分别为100%、91%和70%, 实验结果说明在溶液中游离的·OH对AO7的降解有一定作用.同时反应体系中可能出现单线态氧(1O2), 而糠醇(FFA)通常被认为是1O2的有效猝灭剂, 其反应速率常数为1.2×108 mol-1·s-1[23].当糠醇投加量为10 mmol·L-1时, 30 min反应时间内AO7去除率依旧达到了98%, 说明1O2不是导致AO7降解的主要原因.苯酚(Phenol)对·OH反应速率比叔丁醇更高, 达到了6.6×109 mol-1·s-1[24].不同于叔丁醇的亲水特性, 组成苯酚分子的苯基具有疏水性导致苯酚更加容易聚集在材料表面, 而被当作非均相体系中的自由基清除剂[25].当投加10 mmol·L-1苯酚时, 30 min反应时间内AO7的去除率仅有53%, 反应被极大抑制.从而可以判断AO7的降解主要由·OH完成并且反应主要聚集在Fe3O4-CuO的表面.使用DMPO作为捕获剂捕获反应中的自由基, 使用EPR进行检测, 实验结果如图 10, 观察到了DMPO-·OH加合物(具有aN=aH=14.9G的超精细分裂常数), 进一步证实了Fe3O4-CuO/SPC体系中确实会产生·OH从而降解AO7.但这些自由基猝灭剂均不能完全抑制AO7的降解, 这可能是因为SPC溶于水中产生大量的CO32-和HCO3-离子, 和·OH反应生成CO3-·[26] [式(9)和(10)], 对AO7有一定降解作用[27].

|
(9) |
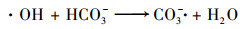
|
(10) |

|
[AO7]0=0.10 mmol·L-1; [Fe3O4-CuO]0=0.2 g·L-1; [SPC]0=2.0 mmol·L-1; pH0=7.0 图 9 不同自由基抑制剂对AO7降解的影响 Fig. 9 Effects of radical quenchers on AO7 degradation |

|
[Fe3O4-CuO]0=0.2 g·L-1; [SPC]0=2.0 mmol·L-1; pH0=7.0 图 10 EPR测试 Fig. 10 Electron paramagnetic resonance experiment |
印染废水一般含有较高的NaCl含量, 其中Cl-对高级氧化反应有较大的影响.图 11可以看出, 随着Cl-投加量的增大, 反应速度明显加快.由表 5可以看出, 一级反应速率常数的增加与Cl-浓度的升高呈线性关系.这可能是因为在Cl-存在的情况下产生了次氯酸或氯气[式(11)~(15)][28~30], 这是一种优秀的偶氮染料漂白剂[31], 从而加快了AO7的氧化降解.

|
(11) |

|
(12) |

|
(13) |

|
(14) |

|
(15) |

|
[AO7]0=0.10 mmol·L-1; [Fe3O4-CuO]0=0.2 g·L-1; [SPC]0=2.0 mmol·L-1; pH0=7.0 图 11 Cl-含量对AO7降解的影响 Fig. 11 Effect of Cl- concentration on AO7 degradation |
|
|
表 5 不同Cl-浓度下的一级反应速率常数 Table 5 Reaction rate constant under different Cl- concentration |
实际生产废水中含有天然有机质(NOM)也会对反应过程产生影响, 在反应体系中添加腐殖酸以模拟实际水体中Fe3O4-CuO/SPC体系对AO7的降解能力.由图 12可知, 当腐殖酸投量分别为10、20和50 mg·L-1时, 30 min内AO7去除率为97%、95%和90%.这主要是因为NOM会消耗反应体系中的自由基, 从而抑制AO7的降解速度.

|
[AO7]0=0.10 mmol·L-1; [Fe3O4-CuO]0=0.2 g·L-1; [SPC]0=2.0 mmol·L-1; pH0=7.0 图 12 HA含量对AO7降解的影响 Fig. 12 Effect of humic acid concentration on AO7 degradation |
在实际运用中, Fe3O4-CuO磁性材料的重复利用性能至关重要, 是衡量活化剂性能的主要指标.为了评价Fe3O4-CuO的重复利用性能, 将反应后的材料使用磁铁与反应液分离并用去离子水和无水乙醇清洗烘干后再次用于AO7降解实验, 结果如图 13.在第2次使用中, 在30 min反应时间内依旧可以达到100%的降解率, 在第3次使用中30 min降解率为97%, 依旧保持了良好的活化性能.在第4次使用中30 min降解率降低到91%.这可能是因为由两个原因导致:一是经过多次使用, 反应中AO7及其降解产物会吸附在材料表面, 降低材料的空隙及比表面积, 从而影响了SPC与活性位点的接触, 降低反应速率[32];二是在反应过程中Fe3O4-CuO材料可能发生离子渗滤从而降低材料的催化能力.取重复利用性实验中第一次反应结束后的反应液经0.22 μm滤头过滤后加入硝酸酸化测量游离金属离子含量.实验结果显示未能检出Fe的渗滤, 而Cu的含量为0.1 mg·L-1, 因此Fe3O4-CuO材料中的Cu的渗滤也是造成催化活性下降的原因之一.

|
[AO7]0=0.10 mmol·L-1; [Fe3O4-CuO]0=0.2 g·L-1; [SPC]0=2.0 mmol·L-1; pH0=7.0 图 13 Fe3O4-CuO重复使用对AO7降解的效果 Fig. 13 Reusability of Fe3O4-CuO for AO7 degradation |
为验证Fe3O4-CuO各组分的活化效果, 分别制备了Fe3O4和CuO进行对比实验, 结果如图 14.当Fe3O4投加量为0.2 g·L-1时, Fe3O4在30 min反应时间内对AO7仅有微弱的去除作用.当投加0.2 g·L-1的CuO时, 在相同时间内对AO7的去除率也仅有49%, 并且这是在CuO含量远远高于投加量Fe3O4-CuO材料中CuO的含量时才能实现.在重复利用性试实验过程中, 检测到反应液中Cu的含量为0.1 mg·L-1, 为了考察渗滤的Cu在反应体系中的作用, 将反应液除去Fe3O4-CuO材料后再次投加AO7与SPC, 实验结果如图 14.从图中可以发现在去除材料的反应液并不能有效活化SPC降解AO7, 这主要是因为材料粒径较小, 反应液中的Cu可能主要以CuO而非Cu离子的形式存在.反应液进入AAS前经硝酸酸化处理后检出Cu离子0.1 mg·L-1的浓度, 但这一浓度的CuO并不能有效活化SPC降解AO7.

|
[AO7]0=0.10 mmol·L-1; [SPC]0=2.0 mmol·L-1; pH0=7.0 图 14 Fe3O4-CuO中各组分对AO7降解的效果 Fig. 14 Effect of Fe3O4-CuO components on AO7 degradation |
本实验结果表明单一的Fe3O4和CuO对AO7的去除效率远不及Fe3O4-CuO, 合成的材料具有显著的协同效果.催化剂的协同效应可能是由于固态的Fe2+还原Cu3+, 推测具体过程如下:固相中的Cu2+与溶液中的双氧水反应生成Cu3+和·OH [式(16)][15], 不稳定的Cu3+和H2O反应继续生成·OH[33] [式(17)]的同时也会迅速的被固态的Fe2+还原为Cu2+从而大大提高了CuO的催化效果.Fe3O4-CuO材料不仅可以利用磁性对材料方便地进行回收利用, 更表现出双金属协同对单独Fe3O4和CuO的催化性能优势, 具有更加广阔的应用前景.

|
(16) |

|
(17) |
为了进一步了解Fe3O4-CuO/SPC体系降解AO7的矿化能力以及反应过程, 进行UV-vis光谱扫描和TOC测试.图 15显示了降解过程中AO7的光谱变化情况.AO7主要含有310、430和484 nm这3个主要的特征峰, 分别对应萘环和偶氮键发色基团[34].随着反应的进行, 484 nm处的吸收峰快速下降, 说明发色基团被快速氧化, 有良好的脱色效果;随着反应时间的延长, 位于310 nm处萘环结构的特征峰也逐渐下降, 说明该体系可以继续氧化AO7降解的中间产物, 具有一定的矿化能力.

|
[AO7]0=0.10 mmol·L-1; [Fe3O4-CuO]0=0.2 g·L-1; [SPC]0=2.0 mmol·L-1; pH0=7.0 图 15 AO7降解过程紫外可见光光谱变化 Fig. 15 Changes in UV-vis spectra during AO7 degradation |
图 16显示了该体系矿化AO7的能力, 随着反应的持续进行, 至60 min时TOC已经由18.79 mg·L-1下降到10.82 mg·L-1, 矿化率达到42.5%.结合光谱扫描推测AO7显色的偶氮键和萘环被氧化后生成以苯环为主体的芳香族化合物, 并且部分中间产物能够进一步降解为小分子有机物并最终矿化为CO2和H2O.

|
[AO7]0=0.10 mmol·L-1; [Fe3O4-CuO]0=0.2 g·L-1; [SPC]0=4.0 mmol·L-1; pH0=7.0 图 16 TOC降解趋势 Fig. 16 TOC removal trends during AO7 degradation |
(1) 采用一步水热法合成了Fe3O4-CuO磁性材料, 该材料可以活化SPC用以降解偶氮染料AO7, 具有良好的脱色效果, 并有较好的矿化作用.
(2) 在Fe3O4-CuO/SPC体系中, AO7的脱色效率随着Fe3O4-CuO的投加量、SPC投加量、初始pH以及Cl-浓度的增大而加快, 但过高的SPC投加量反而会抑制反应的速度.
(3) 降解反应主要由在催化剂表面产生的·OH引起, Fe3O4-CuO中Fe与Cu双金属的氧化还原反应增强了CuO的催化活性, Fe3O4-CuO具有良好的重复利用性.
| [1] | Liu H N, Li G T, Qu J H, et al. Degradation of azo dye acid orange 7 in water by Fe0/granular activated carbon system in the presence of ultrasound[J]. Journal of Hazardous Materials, 2007, 144(1-2): 180-186. DOI:10.1016/j.jhazmat.2006.10.009 |
| [2] | Du C M, Shi T H, Sun Y W, et al. Decolorization of acid orange 7 solution by gas-liquid gliding arc discharge plasma[J]. Journal of Hazardous Materials, 2008, 154(1-3): 1192-1197. DOI:10.1016/j.jhazmat.2007.11.032 |
| [3] | Azam A, Hamid A. Effects of gap size and UV dosage on decolorization of C.I. Acid orange 7 by UV/H2O2 process[J]. Journal of Hazardous Materials, 2006, 133(1-3): 167-171. DOI:10.1016/j.jhazmat.2005.10.005 |
| [4] | Bokare A D, Choi W. Review of iron-free fenton-like systems for activating H2O2 in advanced oxidation processes[J]. Journal of Hazardous Materials, 2014, 275: 121-135. DOI:10.1016/j.jhazmat.2014.04.054 |
| [5] | Wang N N, Zheng T, Zhang G S, et al. A review on fenton-like processes for organic wastewater treatment[J]. Journal of Environmental Chemical Engineering, 2016, 4(1): 762-787. DOI:10.1016/j.jece.2015.12.016 |
| [6] | Ma J, Xia X C, Ma Y, et al. Stability of dissolved percarbonate and its implications for groundwater remediation[J]. Chemosphere, 2018, 205: 41-44. DOI:10.1016/j.chemosphere.2018.04.084 |
| [7] | Fu X R, Gu X G, Lu S G, et al. Benzene depletion by Fe2+-catalyzed sodium percarbonate in aqueous solution[J]. Chemical Engineering Journal, 2015, 267: 25-33. DOI:10.1016/j.cej.2014.12.104 |
| [8] | Fu X R, Gu X G, Lu S G, et al. Benzene oxidation by Fe(Ⅲ)-activated percarbonate:Matrix-constituent effects and degradation pathways[J]. Chemical Engineering Journal, 2017, 309: 22-29. DOI:10.1016/j.cej.2016.10.006 |
| [9] | Li L, Huang J, Hu X B, et al. Activation of sodium percarbonate by vanadium for the degradation of aniline in water:mechanism and identification of reactive species[J]. Chemosphere, 2019, 215: 647-656. DOI:10.1016/j.chemosphere.2018.10.047 |
| [10] | Miao Z W, Gu X G, Lu S G, et al. Enhancement effects of chelating agents on the degradation of tetrachloroethene in Fe(Ⅲ) catalyzed percarbonate system[J]. Chemical Engineering Journal, 2015, 281: 286-294. DOI:10.1016/j.cej.2015.06.076 |
| [11] | Danish M, Gu X G, Lu S G, et al. Effect of solution matrix and pH in Z-nZVI-catalyzed percarbonate system on the generation of reactive oxygen species and degradation of 1, 1, 1-trichloroethane[J]. Water Science & Technology Water Supply, 2017, 17(6): 1568-1578. |
| [12] |
戴竹青, 梁路, 王明新, 等. 纳米四氧化三铁/过碳酸钠降解DDTs及降解产物[J]. 环境科学学报, 2019, 39(4): 1183-1190. Dai Z Q, Liang L, Wang M X, et al. Degradation of DDTs by nano Fe3O4/sodium percarbonate and their degradation products[J]. Acta Scientiae Circumstantiae, 2019, 39(4): 1183-1190. |
| [13] | Danish M, Gu X G, Lu S G, et al. Efficient transformation of trichloroethylene activated through sodium percarbonate using heterogeneous zeolite supported nano zero valent iron-copper bimetallic composite[J]. Chemical Engineering Journal, 2017, 308: 396-407. DOI:10.1016/j.cej.2016.09.051 |
| [14] | Liang H Y, Zhang Y Q, Huang S B, et al. Oxidative degradation of p-chloroaniline by copper oxidate activated persulfate[J]. Chemical Engineering Journal, 2013, 218: 384-391. DOI:10.1016/j.cej.2012.11.093 |
| [15] | Lei Y, Chen C S, Tu Y J, et al. Heterogeneous degradation of organic pollutants by persulfate Activated by CuO-Fe3O4:mechanism, stability, and effects of pH and bicarbonate ions[J]. Environmental Science & Technology, 2015, 49(11): 6838-6845. |
| [16] | Cui H, Gu X G, Lu S G, et al. Degradation of ethylbenzene in aqueous solution by sodium percarbonate activated with EDDS-Fe(Ⅲ) complex[J]. Chemical Engineering Journal, 2017, 309: 80-88. DOI:10.1016/j.cej.2016.10.029 |
| [17] | Qiang Z M, Chang J H, Huang C P. Electrochemical generation of hydrogen peroxide from dissolved oxygen in acidic solutions[J]. Water Research, 2002, 36(1): 85-94. |
| [18] | Buxton C V, Greenstock C L, Helman W P, et al. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (·OH/·O-) in aqueous solution[J]. Journal of Physical and Chemical Reference Data, 1988, 17(2): 513-886. DOI:10.1063/1.555805 |
| [19] | Mezohegyi G, Gonçalves F, Órfão J J M, et al. Tailored activated carbons as catalysts in biodecolourisation of textile azo dyes[J]. Applied Catalysis B:Environmental, 2010, 94(1-2): 179-185. DOI:10.1016/j.apcatb.2009.11.007 |
| [20] | Gould J M. Studies on the mechanism of alkaline peroxide delignification of agricultural residues[J]. Biotechnology and Bioengineering, 1985, 27(3): 225-231. DOI:10.1002/bit.260270303 |
| [21] | Wright P, Abbot J. The oxidation of cinnamaldehyde with alkaline hydrogen peroxide[J]. International Journal of Chemical Kinetics, 1993, 25(11): 901-911. DOI:10.1002/kin.550251104 |
| [22] | Liang C J, Su H W. Identification of sulfate and hydroxyl radicals in thermally activated persulfate[J]. Industrial & Engineering Chemistry Research, 2009, 48(11): 5558-5562. |
| [23] | Yang Z, Jin J, Yuan G, et al. Activation of peroxymonosulfate by benzoquinone:a novel nonradical oxidation process[J]. Environmental Science & Technology, 2015, 49(21): 12941-12950. |
| [24] | Yang S Y, Yang X, Shao X, et al. Activated carbon catalyzed persulfate oxidation of azo dye acid orange 7 at ambient temperature[J]. Journal of Hazardous Materials, 2011, 186(1): 659-666. DOI:10.1016/j.jhazmat.2010.11.057 |
| [25] | Zhang J, Shao X T, Shi C, et al. Decolorization of acid orange 7 with peroxymonosulfate oxidation catalyzed by granular activated carbon[J]. Chemical Engineering Journal, 2013, 232: 259-265. DOI:10.1016/j.cej.2013.07.108 |
| [26] | Li Y, Li L, Chen Z X, et al. Carbonate-activated hydrogen peroxide oxidation process for azo dye decolorization:process, kinetics, and mechanisms[J]. Chemosphere, 2018, 192: 372-378. DOI:10.1016/j.chemosphere.2017.10.126 |
| [27] | Long C, Wei M Y, Huang L H, et al. Efficient H2O2 oxidation of organic dyes catalyzed by simple copper(Ⅱ) ions in bicarbonate aqueous solution[J]. Industrial & Engineering Chemistry Research, 2014, 53(9): 3478-3485. DOI:10.1021/ie403801f |
| [28] | Fei G, Wang L, Li D W, et al. An effective heterogeneous iron-based catalyst to activate peroxymonosulfate for organic contaminants removal[J]. Chemical Engineering Journal, 2015, 267: 102-110. DOI:10.1016/j.cej.2015.01.010 |
| [29] | Huang Y, Wang Z H, Liu Q Z, et al. Effects of chloride on PMS-based pollutant degradation:a substantial discrepancy between dyes and their common decomposition intermediate (phthalic acid)[J]. Chemosphere, 2017, 187: 338-346. DOI:10.1016/j.chemosphere.2017.08.120 |
| [30] | Zhang W Q, Zhou S Q, Sun J L, et al. Impact of chloride ions on UV/H2O2 and UV/persulfate advanced oxidation processes[J]. Environmental Science & Technology, 2018, 52(13): 7380-7389. |
| [31] | Wang Z H, Yuan R X, Guo Y G, et al. Effects of chloride ions on bleaching of azo dyes by Co2+/oxone regent:kinetic analysis[J]. Journal of Hazardous Materials, 2011, 190(1-3): 1083-1087. DOI:10.1016/j.jhazmat.2011.04.016 |
| [32] |
夏文君, 刘锋, 郝尚斌, 等. 石墨烯负载铁锰氧化物活化过一硫酸盐降解金橙G[J]. 环境科学, 2018, 39(5): 2202-2210. Xia W J, Liu F, Hao S B, et al. Degradation of OG with peroxymonosulfate activated by a MnFe2O4-graphene hybrid[J]. Environmental Science, 2018, 39(5): 2202-2210. |
| [33] | Pham A N, Xing G W, Miller C J, et al. Fenton-like copper redox chemistry revisited:Hydrogen peroxide and superoxide mediation of copper-catalyzed oxidant production[J]. Journal of Catalysis, 2013, 301: 54-64. DOI:10.1016/j.jcat.2013.01.025 |
| [34] | Wang K, Zhang J Y, Lou L P, et al. UV or visible light induced photodegradation of AO7 on TiO2 particles:The influence of inorganic anions[J]. Journal of Photochemistry and Photobiology A:Chemistry, 2004, 165(1-3): 201-207. DOI:10.1016/j.jphotochem.2004.03.025 |