 2020, Vol. 41
2020, Vol. 41



2. 南京信息工程大学江苏省大气环境监测与污染控制高技术研究重点实验室, 南京 210044
2. Jiangsu Key Laboratory of Atmospheric Environment Monitoring and Pollution Control, Nanjing University of Information Science & Technology, Nanjing 210044, China
OH自由基(·OH)是大气环境中最重要的氧化剂, 它能通过氧化大气中的挥发性有机物(VOCs)形成过氧自由基, 过氧自由基进一步与氮氧化物(NOx=NO+NO2)反应, 引发一系列的链式反应, 最终导致臭氧和二次有机气溶胶的形成, 而二次有机气溶胶是大气中细粒子(PM2.5)的重要组成部分[1, 2].
在全球范围内, 白天·OH的主要生成机制为臭氧(O3)光解反应, O3受到波长低于319 nm的辐射后光解形成激发态O(1D)原子. O(1D)原子可与水气反应直接生成·OH, 也可能在空气中淬灭返回到基态O(3P)[3]:

|
(1) |

|
(2) |

|
(3) |
·OH的另一个主要来源是甲醛(HCHO)的光解, 波长低于370 nm的辐射, 可使甲醛分解产生H和HCO自由基, H和HCO自由基可以与空气中的O2反应, 然后产生HO2自由基(·HO2), 之后·HO2与NO快速反应形成·OH, HCHO的另一次要光解路径会导致氢气(H2)和一氧化碳(CO)的产生:

|
(4) |
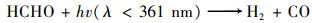
|
(5) |

|
(6) |

|
(7) |

|
(8) |
甲醛作为大气中浓度最高的羰基化合物, 可占羰基化合物总量的70%~80%[4].因此尽管其他醛类也能通过与甲醛光解相类似的反应途径生成·OH, 一般来说甲醛对·OH的贡献在羰基化合物中占主导地位.
许多外场研究表明, 气态亚硝酸(HONO)在不同的环境下都对大气氧化能力有显著影响[5, 6].HONO在辐射波长为300~405 nm范围之间时, 可光解生成·OH:

|
(9) |

|
(10) |
在早期的研究中, HONO浓度在夜间积累并在清晨光解促进大气光化学反应[7].然而, 最近的研究表明, 在白天污染较严重的城市地区, HONO的浓度往往高于预期, 而高浓度的HONO使得其光解不仅在清晨, 甚至在整个白天都是·OH的一个重要来源, 白天可以贡献30%~90%的·OH[5, 8~11].
过氧化氢(H2O2)在波长低于360 nm的辐射下, 同样可直接生成·OH:

|
(11) |
但是H2O2的光解速率较慢, 其水溶性高、大气浓度较低, 对·OH的贡献有限.
与其他生成·OH的反应机制相比, 烯烃与臭氧的反应除白天外, 在夜间同样能生成·OH:

|
(12) |
该反应在化工园区域十分重要.
常州市地处江苏省南部, 为长江三角洲腹地.与长三角其它大都市相比, 常州市更能代表长三角地区的空气污染特征, 对其进行系统观测、研究更能促进对该地区大气污染特征的了解并制定行之有效的污染防治措施.大气氧化性是大气复合污染研究中的一个核心问题, 对大气氧化性的定量描述具有重要意义.为了研究常州大气污染物的变化特征, 尤其是揭示主导该地区污染物形成的大气氧化能力以及找出不同前体物对·OH的贡献, 本研究于2017年春季在常州环境监测中心进行了系统地连续观测实验, 同时提出了对大气氧化能力的定量表征.
1 材料与方法 1.1 采样地点从2017年4月3~24日, 外场观测实验在常州市环境中心(东经119.9°, 北纬31.8°, 图 1)进行.所有仪器放置于配有空调的顶楼, 采样口置于楼顶上方, 高出楼顶约2m, 以防止建筑物表面对采样的干扰.常州环境监测中心位于常州市中心城区, 附近没有明显的工业污染点源, 但交通发达, 车流量较大, 有明显交通高峰期.因此, 周围公路的机动车尾气排放会随气流变化而传送到采样点, 进而造成监测到的污染物浓度的改变.常州环境监测中心内的自动气象站所监测的气象条件能够代表采样点周围的气象条件.

|
图 1 环境监测中心采样点以及常州市地理位置示意 Fig. 1 Map of the sampling site in Changzhou |
本研究中使用的是一套由Ren等[12]研发的基于湿化学方法的气态亚硝酸监测仪.如图 2所示, 首先用磷酸氢二钠作为洗涤剂吸收大气中的亚硝酸, 使之成为亚硝酸盐, 亚硝酸盐衍生形成能高度吸光的偶氮染料(azo dye), 偶氮染料吸收由钨灯光源发射的光, 被液态波导长路径吸收光谱仪检测到其吸光度, 最终计算得出亚硝酸在大气中的浓度.本研究中使用隔膜泵直接抽取环境大气, 空气以1.03L·min-1的流速通过两个串联的5转玻璃线圈采集器.空气流速由质量流量控制器(MSK-M100B)来控制.采样口距离楼顶约1.5m.采样器与放置在室内的亚硝酸检测仪器通过避光聚四氟乙烯管(外径为1.6 nm, 内径为1.0 nm, 长度为7 m)相连. 2 mmol·L-1的磷酸氢二钠缓冲液作为吸收溶液, 用来收集空气样本中的气态亚硝酸.由HONO亨利常数计算可知, 当空气样品流速为1.03L·min-1时, 空气中的气态亚硝酸在经过第一个采样器时被吸收的效率为(99.6±0.5)%.气态亚硝酸从气体溶入吸收液后, 吸收溶液与磺胺(SA)和盐酸萘乙二胺(NED)试剂混合, 在反应线圈(4 m)内发生衍生反应, 亚硝酸盐通过反应式(13)和(14)转化为能高度吸光的偶氮染料.生成的偶氮染料最后通过液体波导毛细管(LWCC, World Precision Instruments)吸收从钨灯光源(HL2000, Ocean Optics)发射的光, 由USB光谱仪(USB4000, Ocean Optics)测定其吸光度, 最终传输为电脑信号值.样品从吸入进样口到最终被检测的滞后时间为(30±0.8)min.

|
图 2 基于湿化学的HONO仪器原理 Fig. 2 Schematics of the custom-built wet chemistry-based HONO instrument |

|
(13) |

|
(14) |
本实验中使用亚硝酸钠标准溶液对亚硝酸测量仪器进行校准.气态亚硝酸在空气样品中的混合比率([HONO])使用以下方程来计算:

|
(15) |
式中, Cl表示吸收液中亚硝酸盐的浓度(mol·L-1), Fl表示吸收液的流速(cm3·min-1), Fg表示空气样品的流速(L·min-1), R为气体常数[8.31 J·(mol·K)-1], T和P分别为大气样品温度(298K)和压强(101 325 Pa), 质量流量控制器在同样的温度和压强下进行校准.
1.2.2 其他测量除了HONO以外, 使用一台自主搭建的质子转移反应-质谱仪(PTR-MS)对包括甲醛在内的部分挥发性有机物(VOCs)进行了同步观测, 具体的仪器介绍参见文献[13]中.本研究中测得的主要烯烃有丙烯、丁烯、异戊二烯和α-蒎烯.同时对痕量污染气体:一氧化碳(CO)(Thermo Scientific, Model 48i)、臭氧(O3)(Thermo Scientific, Model 49i), 二氧化硫(SO2)(Thermo Scientific, Model 43i)和氮氧化物(NOx=NO+NO2)(Thermo Scientific, Model 42i)进行观测.
光解频率则通过光辐射计(德国Meteorology Consult Gmbh)进行测量.光解频率的时间分辨率为1 min, 包括J(O1D)、J(NO2)、J(HONO)、J(H2O2)、J(HCHO)和J(NO3).在模式中使用的其余光解频率由方程式(16)计算得到:

|
(16) |
式中, χ代表太阳天顶角, Li、Mi和Ni是用于晴朗天空条件下的光解参数, 取值来自Jenkin等[14]的研究.计算中通过将理论计算得到的J(NO2)与实际测量得到的J(NO2)进行校准, 最终得到其余未测量的气体在污染条件下的光解频率.
PM2.5数据通过PM2.5在线测量仪器(BAM1020, Met One)得到, 该仪器使用β射线衰减原理自动测量并记录大气环境中的颗粒物质量浓度水平, 时间分辨率为1h.
观测站点位于国家标准气象观测站内, 可以获得实时观测的气象参数, 包括风向、风速、温度、压力和相对湿度.
1.3 模式模拟由于本次观测中没有直接测量·OH的浓度, 本研究使用一个基于光化学机制的MCM (master chemical mechanism)模型[15]来模拟得到·OH.该模型使用FACSIMILE软件(UES Software Inc.)运行.模式中使用的动力学反应速率常数可在MCM网站上获得(http://mcm.leeds.ac.uk/MCM/).在本研究中, 以HONO、O3、NOx、CO、SO2、HCHO等挥发性有机化合物、水蒸气、温度、压力以及光解频率等做为约束条件, 最终得到·OH和·HO2以及其他反应物质的平衡浓度.
本研究中通过蒙特卡罗灵敏度分析以评估模型性能.在每个蒙特卡罗模拟中, 模型的输入变量包括HONO、O3、NO、NO2、CO、SO2、HCHO、VOCs、反应速率常数和光解频率, 每个变量被独立设定在平均值的±10%范围内, 以正态概率分布随机变化.
2 结果与讨论 2.1 观测概况图 3为观测期间部分污染物以及气象参数的时间序列.观测期间温度范围在8.7℃和32.1℃之间, 平均温度18.7℃; 相对湿度在13%和86%之间, 平均相对湿度53%.风速在0.2~4.5 m·s-1范围内变化, 风向以偏西风为主.观测期间, 4月5日夜间开始下雨, 持续到4月6日15:00左右; 4月8日夜间有雨, 持续到4月9日中午.整个观测期间, HONO浓度在0.2~13.9 μg·m-3之间, 平均浓度为(2.9±2.3) μg·m-3, 高于Nie等[16]于2012年春季在南京观测得到的HONO浓度(1.5 μg·m-3), Nie等[16]在南京的观测点为背景站点, 而本研究中观测点受交通源影响较大.图 4为J(HONO)、CO、PM2.5、O3、HCHO、HONO和NOx平均日变化.HONO浓度整体呈现白天浓度低, 夜间浓度高的典型变化趋势, 在早上06:00达到一个峰值, 之后由于太阳光解, HONO浓度开始降低, 直到16:00~17:00浓度达到最低值; 在前半夜, 21:00之后HONO浓度不再增长, 反而略微降低; 在00:00~05:00之间, HONO浓度维持在约4 μg·m-3左右.在观测期间, PM2.5浓度在11~134 μg·m-3之间, 平均浓度(47.9±25.2) μg·m-3; 丙烯(C3H6)、NO、NO2、O3和HCHO的浓度范围分别为:0.1~89.5、0.1~107.0、5.5~184.5、0.3~245.5和0.1~8.8 μg·m-3.观测点的O3与NOx浓度处于较高的水平.CO作为一次排放的示踪物, 其明显的双峰结构表明, 在观测点污染物受交通干道上机动车尾气的直接排放影响较大.NOx的浓度变化趋势也明显受交通源影响, 呈现早晚双高峰的变化趋势.O3浓度的平均日变化则是一个明显的单峰特征, 符合其二次污染物的特点, 约在14:00~15:00左右达到最高值.而观测点的HCHO浓度则没有呈现明显的日变化特征, 这可能是由于HCHO来源既有二次氧化又有机动车的一次排放, 而且二者的强度相当.

|
图 3 2017年4月3~24日风向、风速、温度、相对湿度、C3H6、PM2.5、NO、NO2、O3、HCHO、HONO以及HONO光解频率的时间序列 Fig. 3 Time series of meteorological parameters, including wind direction, wind speed, ambient temperature and relative humidity (RH), mixing ratios of measured C3H6, PM2.5, NO, NO2, O3, HCHO, HONO, and J(HONO) from April 3 to 24, 2017 |

|
误差线表示每小时的标准偏差 图 4 J(HONO)、CO、PM2.5、O3、HCHO、HONO、NOx的平均日变化 Fig. 4 Campaign averaged diurnal profiles of J(HONO), CO, PM2.5, O3, HCHO, HONO, and NOx |
为了评估实验期间常州的大气氧化能力, 本研究利用基于主化学机制(master chemical mechanism, MCM)[17]的盒子模型(box model)模拟了整个观测期间·OH浓度的时间序列(图 5).模型中以HONO、O3、HCHO、NO、NO2、CO、SO2、VOCs、以及水蒸气、温度, 压力和光解频率为约束条件.模拟结果如图 5所示, ·OH的浓度最高值为1.5×107个·cm-3, 白天平均值约为3.4×106个·cm-3, 这一结果与Lu等[18]在广州后花园的观测结果较为接近.为了与太阳辐射强度进行对比, 图 5中还加入了J(HONO)的时间序列, 可见二者的变化趋势十分一致.为了进一步研究二者之间的相关性, 图 5给出了二者观测期间的日平均变化, 二者呈现了显著的相关性, 表明HONO光解可能和·OH的日变化有潜在联系.

|
图 5 MCM模式模拟得到的·OH浓度与HONO光解频率[J(HONO)]的时间序列和平均日变化曲线. Fig. 5 Time series and average diurnal profiles of OH radical concentration simulated by MCM and HONO photolysis frequency [J(HONO)] |
大气中氧化剂的生成和消耗速率决定了大气的氧化能力, 有研究表明, ·OH是白天最主要的氧化剂[19].·OH在大气中基本处于准静态(steady state), 其生成速率与消耗速率近似相等, 因此本研究将·OH的生成速率P·OH定量表征大气氧化能力, 进而分析·OH各来源对大气氧化能力的贡献.
之前的研究表明, HONO光解可以在白天显著增强·OH生成[11, 20].在本研究中分别计算了HONO[方程式(17)]、O3[方程式(18)~(19)]、HCHO[方程式(20)]和H2O2[方程式(21)]的光解以及烯烃的臭氧分解反应[方程式(22)]生成·OH的速率.方程式(17)中的第二项是由于OH+NO形成HONO导致的OH损失[反应式(10)].方程式(17)、(18)、(20)和(21)中, J值是相应物种的光解频率, 方程式(19)中ϕ·OH是O1D与H2O反应而不是被氮气(N2)或氧气(O2)淬灭的部分.计算HCHO光解产生·OH的速率时, 假定由反应式(6)形成的·HO2立即通过反应式(8)转化为·OH.在方程式(22)中, Yield·OH是臭氧和烯烃(i)的气相反应中·OH的产率, kalkene(i)+O3是臭氧与烯烃(i)的反应速率常数.表 1中列出了烯烃的臭氧分解反应的反应速率常数和相应的·OH产率.由于在本研究期间未测量过氧化氢的浓度, 因此在计算中参考了其他研究中给出的观测值(0.7~7.0 μg·m-3)[21~23], 最终采用4 μg·m-3用于计算.
|
|
表 1 计算中使用的挥发性有机化合物(VOCs)的臭氧分解反应速率常数和·OH生成率 Table 1 Ozonolysis reaction rate constants and ·OH formation yields of the volatile organic compounds (VOCs) used in the calculation |

|
(17) |

|
(18) |

|
(19) |

|
(20) |

|
(21) |

|
(22) |
图 6中显示了整个观测期间白天(06:00~18:00)HONO、O3、HCHO和H2O2的光解以及烯烃臭氧化反应对大气氧化能力的贡献.在整个观测期间, O3是最主要贡献者; 而HONO光解对大气氧化能力的贡献在整个观测期间的清晨都占据主要地位, 并在4月5~8日期间的整个白天都有最大贡献.如图 7所示为HONO、O3、HCHO和H2O2的光解以及烯烃臭氧化反应对·OH生成速率以及相对贡献的平均日变化.HONO、O3、HCHO和H2O2光解以及烯烃的臭氧分解产生的平均·OH生成速率为1.13×107、1.98×107、5.07×105、1.58×104和1.35×106个·(cm3·s)-1, 与已有研究值相当[24].可以发现, 在09:00之前, HONO对大气氧化能力的贡献占主导地位; 之后随着O3浓度逐渐增加, O3光解的贡献逐渐增加并在09:00以后开始占主导地位, 直到16:00;在17:00之后由于烯烃浓度的累积, 太阳光照强度下降, 烯烃臭氧化反应对大气氧化性的贡献逐渐上升, 开始占主导地位, 这主要是由于该地区无明显VOC排放源(如化工园区), 烯烃主要来源于机动车排放, 因此在晚高峰期间, 机动车排放较强时, 烯烃与臭氧的反应对·OH生成有明显影响, 而当夜间臭氧迅速消耗后, 该反应对·OH的影响也将减弱.根据蒙特卡罗模拟结果, 白天HONO、O3、HCHO以及烯烃臭氧化反应对·OH生成速率的贡献范围分别为(19.1±4.1)%~(74.9±16.2)%、(7.6±1.0)%~(72.4±11.3)%、(0.4±0.1)%~(1.8±0.3)%和(2.0±0.4)%~(52.7±9.3)%, 平均值分别为(41.1±8.5)%、(46.4±6.8)%、(1.5±0.3)%和(10.9±2.0)%,

|
图 6 观测期间白天(06:00~18:00)HONO、O3、HCHO、H2O2光解以及烯烃臭氧化反应对·OH生成速率贡献的时间序列 Fig. 6 Time series of relative contributions of photolysis of HONO, O3, HCHO, H2O2, and alkene ozonolysis to primary ·OH production during the daytime(06:00-18:00) |

|
误差线代表蒙特卡罗分析的标准偏差 图 7 白天HONO、O3、HCHO、H2O2光解以及烯烃臭氧化反应对·OH的生成速率以及相对贡献的平均日变化 Fig. 7 Average diurnal profiles of daytime production rate and relative contributions of photolysis of HONO, O3, HCHO, H2O2, and alkene ozonolysis to primary ·OH |
H2O2对·OH的生成贡献可忽略不计(0.1%).常州市高浓度的O3是·OH最主要的来源, 而HONO光解对·OH的贡献同样不可忽视.研究表明清晨HONO浓度的提升可促进当天的O3生产并使O3的峰值时间提前[25].受污染物以及天气条件的影响, 各来源对·OH生成的贡献在不同地区各有不同.Elshorbany等[20]于2005年春季在智利圣地亚哥市中心的研究发现, 55%的·OH来源于HONO的光解, 约24%来自烯烃与臭氧的反应, 约16%来自于HCHO光解生成, 5%来自O3光解.Nan等[26]在上海的观测结果表明, HONO、HCHO和O3的光解分别对·OH的贡献为57.6%、30.5%和11.9%, 在该研究中, 采样点受交通源影响较大.Su等[11]于2004年秋季在新垦观测到HONO光解生成·OH的速率约是O3光解的3倍.而Li等[19]于2012年8~12月在中国香港的观测结果则表明O3光解是ROx自由基(·ROx=·OH+·HO2+·RO2)的最主要来源(36%~47%), 其次依次是HONO光解(25%)、HCHO光解(20%~21%), 该研究中观测点在热带气旋的影响下, 在夏秋季节, 常会出现严重的光化学反应引发的烟雾事件.
为了研究颗粒物在光解反应生成·OH时产生的影响, 图 8中分析了不同污染程度下光解频率与·OH之间的相关性.根据空气质量标准, 将PM2.5分为3组, 即 < 35 μg·m-3 (空气质量:优), 35~75 μg·m-3 (空气质量:良), >75 μg·m-3 (空气质量:有污染).J(O1D)为O3光解频率, 与HCHO, HONO的光解频率有一定联系, Ehhalt等[27]在研究中将J(O1D)作为表征这3个前体物的光解频率.在整个观测阶段, ·OH与J(O1D)之间相关性为0.76;同时可以观察到, 随着PM2.5浓度的增加, ·OH与J(O1D)之间线性回归的斜率逐渐降低, 空气质量处于污染状况时, 线性回归的斜率降低更为明显.这可能是当污染物浓度较高时, 削弱了光解作用对·OH的贡献, 降低了·OH的生成速率.Nan等[26]在上海的观测结果同样发现了颗粒物对光解反应生成·OH的削弱作用.当白天污染较严重(PM2.5>75μg·m-3)时, O3的平均浓度为28.5μg·m-3, 低于白天月平均值(51.9μg·m-3), 同时J(O1D)的平均值为5.2×10-6s-1低于月平均值(8.0×10-6 s-1).这也表明颗粒物对光解反应生成·OH的削弱作用可能会进一步对O3浓度产生影响.

|
图 8 白天不同PM2.5浓度下, ·OH与光解频率[J(O1D)]之间的相关性 Fig. 8 Relationship between OH and photolysis rate J(O1D) classified into different PM2.5 levels |
(1) 2017年4月常州环境监测中心附近污染较为严重, 氮氧化物、臭氧以及气态亚硝酸的浓度均处于较高的水平.通过MCM模式模拟, 得到的·OH的浓度最高值为1.5×107个·cm-3, 白天平均值约为3.4×106个·cm-3.
(2) 本研究中提出的·OH生成速率作为表征大气氧化能力指标, 其主要来源对大气氧化性的影响依次为:O3光解(46.4%)>HONO光解(41.1%)>烯烃臭氧化反应(10.9%)>HCHO光解(1.5%)>H2O2光解.
(3) 在清晨时HONO光解对·OH生成起主要作用, 之后随着O3浓度的增加, O3光解对·OH的贡献占主导地位; 在17点之后, 太阳光解频率显著降低后, 烯烃臭氧化反应对·OH生成有主要贡献.白天, 气态亚硝酸和臭氧对·OH的贡献处于比较相当的水平, 即使在下午气态亚硝酸浓度较低时, 它对·OH的贡献依然不可忽视.
致谢: 感谢美国马里兰大学任信荣老师为本研究提供了HONO测量仪器.
| [1] | Turpin B J, Huntzicker J J. Identification of secondary organic aerosol episodes and quantitation of primary and secondary organic aerosol concentrations during SCAQS[J]. Atmospheric Environment, 1995, 29(23): 3527-3544. DOI:10.1016/1352-2310(94)00276-Q |
| [2] |
吕子峰, 郝吉明, 段菁春, 等. 北京市夏季二次有机气溶胶生成潜势的估算[J]. 环境科学, 2009, 30(4): 969-975. Lü Z F, Hao J M, Duan J C, et al. Estimate of the formation potential of secondary organic aerosol in Beijing summertime[J]. Environmental Science, 2009, 30(4): 969-975. |
| [3] | Finlayson-Pitts B J, Pitts J N Jr. Chemistry of the upper and lower atmosphere[M]. San Diego: Academic Press, 1999. |
| [4] | Zheng J, Zhang R Y, Garzón J P, et al. Measurements of formaldehyde at the U.S. -Mexico border during the Cal-Mex 2010 air quality study[J]. Atmospheric Environment, 2013, 70: 513-520. DOI:10.1016/j.atmosenv.2012.09.041 |
| [5] | Zhou X L, Civerolo K, Dai H P, et al. Summertime nitrous acid chemistry in the atmospheric boundary layer at a rural site in New York State[J]. Journal of Geophysical Research:Atmospheres, 2002, 107(D21): 4590. DOI:10.1029/2001JD001539 |
| [6] | Bernard F, Cazaunau M, Grosselin B, et al. Measurements of nitrous acid (HONO) in urban area of Shanghai, China[J]. Environmental Science and Pollution Research, 2016, 23(6): 5818-5829. DOI:10.1007/s11356-015-5797-4 |
| [7] | Platt U, Perner D, Harris G W, et al. Observations of nitrous acid in an urban atmosphere by differential optical absorption[J]. Nature, 1980, 285(5763): 312-314. DOI:10.1038/285312a0 |
| [8] | Neftel A, Blatter A, Hesterberg R, et al. Measurements of concentration gradients of HNO2 and HNO3 over a semi-natural ecosystem[J]. Atmospheric Environment, 1996, 30(17): 3017-3025. DOI:10.1016/1352-2310(96)00011-8 |
| [9] | Kleffmann J, Gavriloaiei T, Hofzumahaus A, et al. Daytime formation of nitrous acid:a major source of OH radicals in a forest[J]. Geophysical Research Letters, 2005, 32(5): L05818. DOI:10.1029/2005GL022524 |
| [10] | Acker K, Möller D, Wieprecht W, et al. Strong daytime production of OH from HNO2 at a rural mountain site[J]. Geophysical Research Letters, 2006, 33(2): L02809. DOI:10.1029/2005GL024643 |
| [11] | Su H, Cheng Y F, Shao M, et al. Nitrous acid (HONO) and its daytime sources at a rural site during the 2004 PRIDE-PRD experiment in China[J]. Journal of Geophysical Research:Atmospheres, 2008, 113(D14): D14312. DOI:10.1029/2007JD009060 |
| [12] | Ren X, Gao H, Zhou X, et al. Measurement of atmospheric nitrous acid at Bodgett Forest during BEARPEX2007[J]. Atmospheric Chemistry and Physics, 2010, 10(13): 6283-6294. DOI:10.5194/acp-10-6283-2010 |
| [13] | Ma Y, Diao Y W, Zhang B J, et al. Detection of formaldehyde emissions from an industrial zone in the Yangtze River Delta region of China using a proton transfer reaction ion-drift chemical ionization mass spectrometer[J]. Atmospheric Measurement Techniques, 2016, 9(12): 6101-6116. DOI:10.5194/amt-9-6101-2016 |
| [14] | Jenkin M E, Saunders S M, Pilling M J. The tropospheric degradation of volatile organic compounds:a protocol for mechanism development[J]. Atmospheric Environment, 1997, 31(1): 81-104. |
| [15] | Saunders S M, Jenkin M E, Derwent R G, et al. Protocol for the development of the Master Chemical Mechanism, MCM v3(Part A):tropospheric degradation of non-aromatic volatile organic compounds[J]. Atmospheric Chemistry and Physics, 2003, 3(1): 161-180. |
| [16] | Nie W, Ding A J, Xie Y N, et al. Influence of biomass burning plumes on HONO chemistry in eastern China[J]. Atmospheric Chemistry and Physics, 2015, 15(3): 1147-1159. DOI:10.5194/acp-15-1147-2015 |
| [17] | Jenkin M E, Wyche K P, Evans C J, et al. Development and chamber evaluation of the MCM v3.2 degradation scheme for β-caryophyllene[J]. Atmospheric Chemistry and Physics, 2012, 12(11): 5275-5308. DOI:10.5194/acp-12-5275-2012 |
| [18] | Lu K D, Rohrer F, Holland F, et al. Observation and modelling of OH and HO2 concentrations in the Pearl River Delta 2006:a missing OH source in a VOC rich atmosphere[J]. Atmospheric Chemistry and Physics, 2012, 12(3): 1541-1569. DOI:10.5194/acp-12-1541-2012 |
| [19] | Li Z Y, Xue L K, Yang X, et al. Oxidizing capacity of the rural atmosphere in Hong Kong, Southern China[J]. Science of the Total Environment, 2018, 612: 1114-1122. DOI:10.1016/j.scitotenv.2017.08.310 |
| [20] | Elshorbany Y F, Kurtenbach R, Wiesen P, et al. Oxidation capacity of the city air of Santiago, Chile[J]. Atmospheric Chemistry and Physics, 2009, 9(6): 2257-2273. DOI:10.5194/acp-9-2257-2009 |
| [21] | Hua W, Chen Z M, Jie C Y, et al. Atmospheric hydrogen peroxide and organic hydroperoxides during PRIDE-PRD'06, China:their concentration, formation mechanism and contribution to secondary aerosols[J]. Atmospheric Chemistry and Physics, 2008, 8(22): 6755-6773. DOI:10.5194/acp-8-6755-2008 |
| [22] | Ren Y, Ding A J, Wang T, et al. Measurement of gas-phase total peroxides at the summit of Mount Tai in China[J]. Atmospheric Environment, 2009, 43(9): 1702-1711. DOI:10.1016/j.atmosenv.2008.12.020 |
| [23] | Guo J, Tilgner A, Yeung C, et al. Atmospheric peroxides in a polluted subtropical environment:seasonal variation, sources and sinks, and importance of heterogeneous processes[J]. Environmental Science & Technology, 2014, 48(3): 1443-1450. |
| [24] | Alicke B, Platt U, Stutz J. Impact of nitrous acid photolysis on the total hydroxyl radical budget during the Limitation of Oxidant Production/Pianura Padana Produzione di Ozono study in Milan[J]. Journal of Geophysical Research:Atmospheres, 2002, 107(D22): 8196. DOI:10.1029/2000JD000075 |
| [25] | Li G, Lei W, Zavala M, et al. Impacts of HONO sources on the photochemistry in Mexico City during the MCMA-2006/MILAGO Campaign[J]. Atmospheric Chemistry and Physics, 2010, 10(14): 6551-6567. DOI:10.5194/acp-10-6551-2010 |
| [26] | Nan J L, Wang S S, Guo Y L, et al. Study on the daytime OH radical and implication for its relationship with fine particles over megacity of Shanghai, China[J]. Atmospheric Environment, 2017, 154: 167-178. DOI:10.1016/j.atmosenv.2017.01.046 |
| [27] | Ehhalt D H, Rohrer F. Dependence of the OH concentration on solar UV[J]. Journal of Geophysical Research:Atmospheres, 2000, 105(D3): 3565-3571. DOI:10.1029/1999JD901070 |