 2019, Vol. 40
2019, Vol. 40

2. 上海市环境科学研究院, 国家环境保护城市大气复合污染成因与防治重点实验室, 上海 200233
 , QIAO Li-ping2 , LOU Sheng-rong2 , YAN Ru-sha2 , ZHOU Min2 , LIU Yu-cun2 , FENG Jia-liang1
, QIAO Li-ping2 , LOU Sheng-rong2 , YAN Ru-sha2 , ZHOU Min2 , LIU Yu-cun2 , FENG Jia-liang1 , HUANG Dan-dan2
, HUANG Dan-dan2
2. State Environmental Protection Key Laboratory of Formation and Prevention of the Urban Air Complex, Shanghai Academy of Environmental Sciences, Shanghai 200233, China
大气中的细颗粒物不仅危害人体健康, 容易引发呼吸道和心血管疾病, 还能够吸收和散射太阳辐射、降低能见度, 对全球气候有着重要影响[1, 2].细颗粒物的来源主要包括自然源(如矿物粉尘和海盐)和人为源(如交通、燃烧、工业和农业活动等)[3, 4].有研究表明直径1 μm以下的颗粒物(即亚微米颗粒物, PM1)多是由挥发性有机物(volatile organic compounds, VOCs)在大气中经过二次反应生成的; 与PM2.5相比, PM1由于粒径较小、在大气中停留时间长, 可传输较远的距离; 且其比表面积大, 易吸附大量有害物质对健康的影响比PM2.5更显著.此外, PM1颗粒物的等效直径大小与太阳短波辐射的波长最为接近, 因此其消光作用最强, 是导致污染期间能见度快速下降的主要原因[5, 6].因此, 开展对PM1颗粒物的研究成为大气气溶胶领域的焦点.
PM1中的主要化学组分包括有机气溶胶(organic aerosol, OA)以及水溶性无机组分(如硫酸盐、硝酸盐、铵盐和氯离子).无机组分中, 硫酸盐(SO42-)、硝酸盐(NO3-)和铵盐(NH4+)主要来源于气态前体物(SO2、NOx和NH3)的二次转化, 通常称为二次无机气溶胶(secondary inorganic aerosol, SIA). Zhang等[7]对全球37个站点PM1中的化学组分分析表明:二次无机气溶胶(SIA=SO42-+NO3-+NH4+)在非难熔性亚微米气溶胶中(NR-PM1, 不包含黑碳)平均贡献为55%. OA是PM1最复杂的组成部分, 约占到细颗粒物质量浓度的20%~90%[8]; 主要包括由污染源直接排放的一次有机气溶胶(POA)、一次排放的气态有机污染物在大气中经过进一步化学反应而生成的二次有机气溶胶(SOA)以及大气中的VOCs与氧化剂(如OH、O3和NO3)发生光化学反应后, 生成半挥发和难挥发性有机物, 经过气-粒分配凝结到已有颗粒物上形成的SOA[9].相比于POA, SOA具有很强的吸湿性和较高的溶解度, 对气溶胶的光学辐射特性和健康效应等有更大影响.全球模型结果表明, 在中纬度地区, SOA约贡献有机物浓度的50%[10].在中国, 颗粒物中的SOA约占总有机物浓度的30%~95%[11].综上所述, 二次气溶胶(SIA+SOA)是PM1的最主要构成部分.但由于目前对PM1中二次气溶胶的化学组成与性质、氧化性以及来源转化机制缺乏全面认识, 因此, 对二次气溶胶的研究仍然不够深入.
以往研究大多数都是基于滤膜采样, 受化学分析所需样品量及人工操作的限制, 时间分辨率低[12], 无法捕捉到气溶胶的快速演化过程; 基于在线Marga或者OC/EC的数据, 时间分辨率也较低, 并且只能根据经验公式对二次有机组分进行估算[13, 14], 偏差较大, 无法实现对二次有机组分的直接定量.近年来高分辨率在线测量仪器——气溶胶质谱仪(AMS)的出现大大推动了大气气溶胶来源解析研究方面的发展, 其可以更好地解释环境二次气溶胶的化学特征、来源以及形成过程.但我国对于AMS的应用与欧美等发达国家相比起步较晚, 关于上海地区高时间分辨率亚微米气溶胶特性的研究工作更是寥寥无几, 无法洞悉上海地区二次气溶胶的形成、变化特点以及季节变化规律.因此有必要针对区域污染地区开展长期、连续、高时间分辨率的观测研究工作, 从而深入研究上海城区PM1中二次气溶胶的形成机制与主要影响因素.
本研究利用高分辨率飞行时间气溶胶质谱仪(HR-TOF-AMS)对上海城区2018年春季及夏季的亚微米颗粒物(PM1)进行实时在线观测, 通过了解上海大气中亚微米颗粒物的污染状况, 厘清污染成因, 以期为未来颗粒物的治理提供科学依据.
1 材料与方法 1.1 采样地点和时间观测地点位于上海市环境科学研究院(SAES, 31.17°N, 121.43°E), 该测点地处上海中心城区西南部的徐汇区, 周围被商业区和住宅包围, 主要受到机动车尾气排放、餐饮烹饪排放、局地二次反应生成以及区域传输的影响, 基本代表了上海市城区的空气质量状况[15].采样口架设于一座五层建筑物的屋顶, 距地面约15 m高.此次观测时间为2018年4月10~25日(春季)、2018年8月21日~9月14日(夏季).
1.2 监测仪器PM1颗粒物的粒径及各组分质量浓度由高分辨率飞行时间气溶胶质谱仪(HR TOF AMS, AMS)测定.颗粒物经由一个PM2.5(流量3 L·min-1)旋风切割头后, 经过Nafion扩散除湿管除湿, 使相对湿度稳定在30%左右, AMS进样口流速为80 mL·min-1.在本次观测中, AMS采样由V模式和W模式交替进行, 每2 min扫描一次.其中, V模式具有较高的信号强度, 用于获得颗粒物各化学组成的质量浓度; 而W模式具有较高的质谱分辨率, 用于有机物的源解析.观测开始和结束时, 分别对AMS的流量、电离效率及粒径进行标定. PM2.5质量浓度采用Thermo Fisher Scientific Inc, FH62C-14颗粒物监测仪测定, 时间分辨率为5 min, 样品流量是16.7 L·min-1.有机碳(OC)和元素碳(EC)浓度采用OC/EC分析仪测量(Sunset Laboratory Inc, RT-4), 该仪器采用热/光吸收原理, 时间分辨率为60 min; 气相污染物(NO-NO2-NOx和O3)分别由Thermo scientific 42i NO-NO2-NOx分析仪(仪器精度为±7%)、Ecotech EC9810B O3分析仪测定, 最低检测限为0.5×10-9, 时间分辨率为5min.
1.3 AMS数据处理使用基于Wave Metrics Igor Pro开发的AMS标准数据分析软件包SQUIRREL version 1.61和PIKA version 1.21分别处理V模式(unit mass resolution, UMR, 单位质量数分辨率质谱)和W模式(high resolution, HR, 高分辨率质谱)对应的有机物质谱数据, 从而获得有机气溶胶的质量浓度以及元素组成等信息.数据处理方法:Cl-、NO3-、SO42-以及OA的相对电离效率(relative ionization efficiency, RIE)分别取1.3、1.1、1.2和1.4[16, 17], 而NH4+的RIE使用的是校正值4.05.在计算质量浓度时, 考虑到颗粒物在热蒸发器上的反弹以及传输效率等造成的损失, 颗粒与离子碎片的收集效率(collection efficiency, CE)采用经验值0.5[18].
随后利用正交矩阵因子解析模型(positive matrix factorization, PMF)对2018年春季和夏季有机组分进行源解析. PMF源解析模型需要有机物离子碎片的浓度矩阵和误差矩阵, 误差矩阵按照Ulbrich等[19]描述的方法使用PMF评估工具包(pmf evaluation toolkit, PET)进行预处理.即:删除信噪比(SNR)小于0.2的变量(“坏”变量), 而对于信噪比在0.2~2之间的变量(“弱”变量)将其误差扩大2倍.由于离子碎片O+、HO+、H2O+和CO+仅与CO2+相关, 因此将这些m/z中每一个的误差值乘以sqrt(5)以避免过度加权CO2+.在PMF2算法运行结束后, 根据Zhang等[20]所描述的方法评估PMF运行后的结果.本文中, PMF分析中所有数据集的fpeak值取0, 对于有机物因子数的识别是依靠拟合残差大小、Q/Qexp值、质谱图特征、元素组成、OM/OC、日变化规律以及与示踪物的相关性等信息进行判断.
此外, AMS对各组分浓度进行定量的误差为30%[21].误差主要来自电离效率、采样流量、颗粒物收集效率及其在透镜中的透过率等4个方面. 4个不确定因素对颗粒相中各组分的影响相同, 不影响本文中相对贡献的探讨.
2 结果与讨论 2.1 PM1颗粒物中主要化学组成本次观测期间上海城区春季和夏季PM1的主要化学组成、浓度水平、气象条件以及PM2.5质量浓度的时间序列如图 1所示.观测期间春季和夏季上海城区的平均温度分别为18.5℃和32.5℃; 平均湿度为63.8%和75.6%;平均风速为4.5 m·s-1和4.5 m·s-1.整个观测期间, PM1和PM2.5的变化趋势基本一致.春季PM1和PM2.5的平均质量浓度分别为21.5μg·m-3和36.4μg·m-3, PM1的质量浓度显著低于2010年上海世博会期间的PM1平均浓度(27.2 μg·m-3)[17]; 夏季PM1和PM2.5的平均质量浓度分别为15.6μg·m-3和21.7μg·m-3, PM1的质量浓度显著低于2012年长三角区域的PM1平均浓度(29.9μg·m-3)[22].部分原因是由于上海地区2018年夏季受西北太平洋台风的影响, 台风偏多, 且强度较大, 导致PM1浓度处于较低水平.但总体而言, 近期的观测结果表明上海近些年空气质量有了显著的改善.春季和夏季PM1分别占PM2.5质量浓度的59%和72%, 夏季PM1对PM2.5的贡献远高于春季, 可能是由于夏季光化学作用强烈, 有大量的SO42-及SOA生成, 增加了细粒子的质量浓度, 本研究会在下文进行进一步地讨论; 相比之下, 春季PM1在PM2.5中的占比较低, 可能是由于受到春季沙尘天气的影响[23](观测期间春季和夏季PM10质量浓度分别为82.1μg·m-3和32.7μg·m-3; PM2.5/PM10的比值分别为0.4和0.7).

|
图 1 春季和夏季不同区间内PM1主要化学组分浓度以及气象条件的时间序列 Fig. 1 Time series of major PM1 component concentrations and meteorological parameters in spring and summer |
整个观测期间, PM1颗粒物中主要成分依次为OA、SO42-、NO3-、NH4+和Cl-. OA是PM1的重要组成部分, 春季和夏季平均浓度分别为12.7μg·m-3和7.9μg·m-3, 对PM1的平均贡献分别为59%和51%, 显著高于何瑶等[24]统计的2008年以来长三角区域有机物对PM1贡献的35%~51%;春季和夏季二次无机组分(SIA)平均浓度分别为8.0μg·m-3和7.5μg·m-3, 对PM1的平均贡献分别为40%和48%, 低于Zhang等[7]统计全球37个站点的结果(SIA占PM1的55%).近年来, 随着对SO2和NOx等无机气态污染物排放控制的加强, 颗粒相无机组分的浓度降低, 可能是导致颗粒相中有机组分相对贡献上升的直接因素; 此外, 近年来大气氧化性逐渐增强, O3及Ox(O3+NO2)浓度上升, 可能导致颗粒相中二次有机物浓度上升, 从而进一步增加有机物对PM1的贡献.
2.2 光化学氧化和液相反应对二次无机气溶胶形成的影响春季观测期间, PM1质量浓度的时间序列与Ox(氧化剂)相关性较好, 表明光化学氧化可能对春季二次颗粒物的生成有较大的影响, 尤其以4月17~23日(EP1)的局地光化学氧化过程最显著, PM1与Ox的相关性可以达到0.6; EP1主要受偏东风清洁气流的影响, 风速较大(平均风速4.5 m·s-1), 受区域传输的影响较小; 在此期间局地温度、湿度也不断攀升(温度由22℃上升至26℃, 湿度由23%上升至94%).夏季8月14~31日(EP2)的过程中, PM1质量浓度的时间序列也与Ox有较好的一致性(R2=0.7);而在9月4~14日(EP3)的过程中, 湿度较高(平均70.6%)且风速较小(平均风速3.3 m·s-1), 尤其是9月4~8日, 颗粒物中NO3-浓度增长显著, 可能受到液相反应过程的影响.因此, 下文讨论中以EP1和EP2为特征时段, 研究春季和夏季光化学氧化过程对二次气溶胶生成的影响; 以EP3为特征时段研究液相反应对二次气溶胶生成的影响.
EP1与春季其他时段相比, PM1平均质量浓度相差不大(分别为21.7μg·m-3和21.4μg·m-3), 但SO42-占比由21.6%上升至28.7%[图 1(a)]; 同时通过春季的日变化曲线可以看出, EP1中O3[图 2(c)]和SO2[图 2(b)]与春季其他时段相比, 浓度值明显升高, 且SO42-浓度[图 2(a)]在正午12:00~14:00左右有显著的上升, 质量浓度约为其他时段的1.5倍.为进一步研究气相氧化转化对SO42-的贡献, 本研究计算了SO2向颗粒相SO42-的二次转化率(SOR):

|
图 2 春季不同区间内SO42-、SO2、O3以及SOR日变化 Fig. 2 Diurnal profiles of SO42-, SO2, O3, and SOR during different periods in spring |
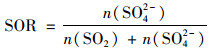
|
SOR可以清楚地指示SO2的二次转化过程[25, 26].有研究认为[27, 28], 当SOR>0.1, 表明大气中有光化学氧化过程发生; SOR值越大, 气相SO2发生光化学氧化反应转化为SO42-的几率越大. EP1中, SOR在正午时段(12:00~14:00)出现明显上升[图 2(d)], 显著高于其他时段正午的峰值. SO42-是热稳定的物质, 正午时段氧化剂浓度与温度较高, 光化学活动活跃, 有利于SO2发生光化学氧化反应转化为SO42-[29, 30].由此可见, 在不受污染气流影响时, 春季的光化学氧化过程对SO42-的生成有显著贡献, 导致SO42-在PM1中的占比上升显著.
对比夏季的2个二次气溶胶形成过程, 本研究发现在夏季液相反应过程中(EP3, 19.4μg·m-3)PM1的平均质量浓度显著高于光化学氧化过程(EP2, 11.7μg·m-3).液相反应过程中OA、SO42-、NO3-、NH4+和Cl-的质量浓度分别是光化学氧化过程的1.5、1.4、3.4、1.9与1.6倍.夏季液相反应过程中, NO3-质量浓度上升最为显著, 在PM1中的占比由8%上升至16%[图 1(a)].为进一步探究影响NO3-生成的主要因素, 本文利用在线气溶胶热力学模型E-AIMII对颗粒相中的水含量(LWC, μg·m-3)进行计算[31].发现液相反应期间, 颗粒相水含量与NO3-质量浓度有较好的一致性, 相关性系数达到了0.7[图 3(e)].且液相反应过程中, NO3-在夜间出现峰值[图 3(a)], 与颗粒相水含量的日变化趋势一致.夏季O3浓度较高, 有利于夜间NO3-的前体物N2O5的生成[32]; 而夜间的高湿环境进一步促进了液相反应的发生, 即:气态N2O5溶解在潮湿颗粒物及悬浮小液滴表面的液相环境后, 迅速发生非均相水解反应, 生成HNO3.以上结果表明, 夜间发生的液相反应对夏季NO3-的“暴发性增长”有较大的促进作用.

|
图 3 夏季不同区间内NO3-、O3、NO2与颗粒相水含量日变化特征以及颗粒相水含量时序 Fig. 3 Diurnal profiles of NO3-, O3, NO2, and liquid water contents and time series of aerosol liquid water contents during different periods in summer |
如2.1节所描述, OA是PM1的重要组成部分.为了深入分析有机气溶胶的来源及其形成的主要影响因素, 本研究首先利用PMF对AMS得到的春季和夏季高分辨率有机物质谱数据进行源解析, 研究POA与SOA的相对贡献; 随后对POA与SOA的成因进行了分析. PMF解析因子的时间序列、指纹谱、元素组成(O/C、H/C)、有机物与有机碳的比值(OM/OC)以及日变化规律如图 4~6所示.春季和夏季识别出四类有机气溶胶因子分别为:还原性有机气溶胶(HOA), 餐饮源有机气溶胶(COA), 新鲜生成的(半挥发)低氧化性有机气溶胶(SV-OOA)和老化的(低挥发)高氧化性有机气溶胶(LV-OOA).根据元素组成以及指纹谱的特征, 其中春季解析出的高氧化性有机气溶胶LV-OOA又可以分为LV-OOA1(O/C=0.7; OM/OC=2.1)和LV-OOA2(O/C=1.0; OM/OC=2.4)两类; 夏季解析出的低氧化性有机气溶胶SV-OOA可再分为SV-OOA1(O/C=0.2; OM/OC=1.5)和SV-OOA2(O/C=0.3; OM/OC=1.6)两类.

|
图 4 春季PMF解析得到的5个因子的时间序列以及指纹图谱 Fig. 4 Time series and mass spectra of PMF-resolved factors in spring |

|
图 5 夏季PMF解析得到的5个因子的时间序列以及指纹图谱 Fig. 5 Time series and mass spectra of PMF-resolved factors in summer |

|
图 6 PMF解析不同类型的有机物在春季和夏季的日变化曲线 Fig. 6 Diurnal profiles of PMF-resolved organic factors in spring and summer |
POA具有氧化程度较低、受源排放直接影响的特点.在此次观测过程中, 笔者识别了两类一次有机气溶胶, 即还原性有机气溶胶(HOA)与餐饮源有机气溶胶(COA). HOA主要来自化石燃料燃烧时的直接排放, 其源谱主要由燃料中的CnH2n+1+和CnH2n-1+等烷烃类离子碎片构成(如C3H7+ m/z=43; C4H7+ m/z=55; C4H9+ m/z=57等)[8], 其在HOA图谱中约占到55%[33].春[图 4(a)]、夏[图 5(a)]季HOA的时间序列均与一次燃烧源排放的EC具有较好的相关性, 并且氧化程度较低(春季和夏季HOA的O:C、H:C分别为0.2、1.9; 0.3、1.9);在早上07:00~09:00以及晚上18:00~20:00出现峰值, 与上、下班早、晚高峰期一致[图 6(a)和6(f)], 表明机动车尾气排放是HOA的重要来源.观测期间上海城区春季和夏季HOA平均质量浓度分别为1.7μg·m-3和0.9μg·m-3, 对总有机物的贡献分别为12.2%和14.7%, 可见城区机动车尾气对颗粒相有机物的贡献总体而言较为稳定.
COA主要来自于餐饮油烟[34], 其特征峰为C3H3O+(m/z=55)、C6H10O+(m/z=98), 并且COA中C4H7+/C4H9+的比值约是HOA的2.5倍[8].本次观测中解析出的春[图 4(a)]、夏[图 5(a)]季COA的时间序列与其特征峰C6H10O+的时间序列具有较高的一致性, 同时其日变化曲线[图 6(b)和6(g)]呈现明显的双峰型, 峰值出现在中午12:00以及晚上19:00, 与用餐时间相吻合, 说明此因子确实代表餐饮油烟排放.此外, 从COA的日变化曲线可以看出:春季和夏季不同污染过程中, COA的质量浓度以及日变化趋势均较为稳定, 浓度变化不大, 不受传输、气象条件及大气氧化性的影响.与机动车源类似, 在上海典型城区点位, 餐饮油烟对局地贡献也较为稳定(平均浓度~2.4μg·m-3, 占总有机物的20%).相关研究也表明:COA主要来自人口密集的城市地区, 平均贡献到总有机物的20%(14%~25%), 是城市有机物中较为稳定的组分[35].观测期间上海城区春季和夏季COA对POA的贡献占到63%以上, 表明餐饮油烟在上海城区一次源排放的污染物中具有稳定且显著的贡献.
总体而言, 上海城区一次有机污染物(即POA=HOA+COA)对总有机物的贡献为34%, 占PM1颗粒物质量浓度的18.2%, 主要受到机动车尾气及餐饮排放的影响.
2.3.2 光化学氧化和液相反应对SOA形成的影响根据SOA的老化程度, SOA又可以划分为SV-OOA和LV-OOA.其中, LV-OOA的氧化程度高于SV-OOA, 如高氧化态有机酸的特征峰CO2+在LV-OOA图谱中的贡献为17%~21%, 而在SV-OOA中的贡献仅为7%~14%[8].
春季LV-OOA可进一步解析出两个来源, 即LV-OOA1与LV-OOA2[图 4(a)]. LV-OOA2与LV-OOA1相比, 氧化程度较高, 相对分子质量较大的离子碎片含量低[图 4(b)](LV-OOA1与LV-OOA2中m/z>80的离子碎片贡献分别为0.1、0.1).其中在春季EP1中受光照强度、氧化剂浓度等因素的影响, SV-OOA、LV-OOA1与LV-OOA2均在正午时段有显著生成[图 6(c)~6(e)].尤其是LV-OOA1, 日间浓度显著高于夜间浓度, 在14:00左右达峰值, 表明局地光化学氧化过程也会促进低挥发性有机物的生成, 与香港的观测结果相似[36]; EP1期间LV-OOA1在SOA中的贡献高达59%.而春季除EP1外的其他时段, LV-OOA1浓度较高但日变化趋势波动不大.此外, 春季解析出的高氧化态LV-OOA2在春季两个不同区间内, 日变化趋势较稳定(日浓度变化范围<1 μg·m-3), 且两个区间内质量浓度无显著差异, 表明LV-OOA2可能来自区域传输, 可认为是城市背景中较为老化的气溶胶, 占二次有机气溶胶的24.6%. EP1时段, SV-OOA与NO3-的相关性较差[图 4(a)], 且在正午小幅上升; 而其他时段, SV-OOA于清晨06:00达峰值, 可能是受残留层化学的影响, 即清晨边界层打开, 残留层污染物回流到对流层发生氧化转化引起[37, 38]; 随后受温度和边界层抬升的双重影响, SV-OOA浓度持续下降, 并在下午出现最低值; 到傍晚18:00之后, 随着温度降低, 相对湿度升高, SV-OOA浓度增加[图 6(c)], 与NO3-相关性较好.
夏季SV-OOA可进一步解析出两个来源, 即SV-OOA1和SV-OOA2[图 5(a)]. SV-OOA2与SV-OOA1相比, 氧化程度较高; 相对分子质量较大的离子碎片因官能团中含有较多的氧, 在图谱中表现为大分子量的离子碎片含量较高[图 5(b)](SV-OOA1与SV-OOA2中m/z>80的离子碎片贡献分别为0.2和0.3).夏季SV-OOA1、SV-OOA2与LV-OOA均在正午时段出现峰值[图 6(h)~6(j)]. EP2(光化学氧化)中SV-OOA2在正午时段上升显著, 与Ox相关性较好[图 5(a)], 表明SV-OOA2可能是局地光化学的产物, 其质量浓度约是EP3(液相过程)中SV-OOA2的9倍[图 6(i)]. EP3过程中, SV-OOA1与LV-OOA均在夜间出现第2个峰值, 与颗粒相水含量有一定的相关性, 表明液相反应中进一步促进了夜间SOA的生成.
此外, 本研究估算了光化学氧化过程以及液相反应对SOA的贡献的最大值.即, 分别对正午时段以及夜间SV-OOA和LV-OOA的平均浓度进行积分, 随后计算出其在相应过程中所占的比重, 从而估算出春季和夏季观察到的3个过程对SOA的贡献.对比春季和夏季出现的两次光化学氧化过程(EP1和EP2)发现:本次观测期间春季和夏季光化学氧化过程对SOA的贡献分别为28.2%和40.5%, 夏季温度高、光化学反应活跃,加之较高的VOCs浓度[39, 40], 有利于SOA的生成, 因此表现为夏季光化学氧化过程对SOA的贡献显著大于春季; 而夏季夜间的液相反应(EP3)对SOA的贡献为33.3%.
综上所述, 本次观测期间春季SOA(即SOA=SV-OOA+LV-OOA)的生成主要受到光化学氧化的影响; 而夏季污染期SOA的生成受到光化学氧化和液相反应的共同影响.总体而言, SOA对总有机物的贡献为66%, 对PM1颗粒物的贡献为35.5%.相比于二次无机物(PM1中占比43%), 二次有机物已成为影响上海空气质量的主要因素, 加强气相有机活性前体物的管控对未来上海空气质量的改善至关重要.
3 结论(1) 本研究利用高分辨率飞行时间气溶胶质谱仪对上海城区春季和夏季的亚微米颗粒物进行实时在线表征.春季和夏季PM1平均质量浓度分别为22.4μg·m-3和15.6μg·m-3.其中有机物是PM1的重要组成部分, 对PM1的平均贡献分别为59%和51%.
(2) 观测期间, 春季和夏季共观察到2个光化学氧化过程和1个液相反应过程, 其中春季光化学氧化期间SO42-和老化的气溶胶在正午时段上升显著; 夏季观察到的液相反应过程中, NO3-质量浓度约为夏季光化学氧化过程的3倍且SOA在夜间出现第二个峰值, 表明夜间发生的非均相反应对NO3-的“暴发性增长”以及SOA的生成有较大的贡献.
(3) 四类有机气溶胶:HOA、COA、SV-OOA和LV-OOA在不同季节对总有机物的平均贡献为13.4%、20.6%、37.5%和28.6%.其中一次、二次有机气溶胶对总有机物的贡献分别为34%和66%, 实现了对POA和SOA的直接定量.
(4) 春季光化学氧化过程对SOA的贡献为28.2%, 其中对氧化程度高的LV-OOA1贡献最为显著, 高达41.7%;夏季光化学氧化过程对SOA的贡献为40.5%, 其中对氧化程度略低的SV-OOA2贡献显著, 高达38.7%;而夏季夜间的液相反应对SOA的贡献为33.3%.
| [1] | Qiao L P, Cai J, Wang H L, et al. PM2.5 constituents and hospital emergency-room visits in Shanghai, China[J]. Environmental Science & Technology, 2014, 48(17): 10406-10414. |
| [2] | Chow J C, Watson J G, Kuhns H, et al. Source profiles for industrial, mobile, and area sources in the big bend regional aerosol visibility and observational study[J]. Chemosphere, 2004, 54(2): 185-208. DOI:10.1016/j.chemosphere.2003.07.004 |
| [3] | May A A, Nguyen N T, Presto A A, et al. Gas-and particle-phase primary emissions from in-use, on-road gasoline and diesel vehicles[J]. Atmospheric Environment, 2014, 88: 247-260. DOI:10.1016/j.atmosenv.2014.01.046 |
| [4] | Cheng Z, Wang S, Fu X, et al. Impact of biomass burning on haze pollution in the Yangtze River delta, China:a case study in summer 2011[J]. Atmospheric Chemistry and Physics, 2014, 14(9): 4573-4585. DOI:10.5194/acp-14-4573-2014 |
| [5] |
林瑜, 叶芝祥, 杨怀金, 等. 成都市中心城区大气PM1的污染特征及来源解析[J]. 中国环境科学, 2017, 37(9): 3220-3226. Lin Y, Ye Z X, Yang H J, et al. Pollution level and source apportionment of atmospheric particles PM1 in downtown area of Chengdu[J]. China Environmental Science, 2017, 37(9): 3220-3226. DOI:10.3969/j.issn.1000-6923.2017.09.003 |
| [6] | Seinfeld J I. Atmospheric chemistry and physics:from air pollution to climate change[J]. Environment:Science and Policy for Sustainable Development, 1998, 40(7): 26. |
| [7] | Zhang Q, Jimenez J L, Canagaratna M R, et al. Ubiquity and dominance of oxygenated species in organic aerosols in anthropogenically-influenced Northern Hemisphere midlatitudes[J]. Geophysical Research Letters, 2007, 34(13): L13801. |
| [8] | Hu W, Hu M, Hu W W, et al. Seasonal variations in high time-resolved chemical compositions, sources, and evolution of atmospheric submicron aerosols in the megacity Beijing[J]. Atmospheric Chemistry and Physics, 2017, 17(16): 9979-10000. DOI:10.5194/acp-17-9979-2017 |
| [9] | Chapleski R C, Zhang Y F, Troya D, et al. Heterogeneous chemistry and reaction dynamics of the atmospheric oxidants, O3, NO3, and OH, on organic surfaces[J]. Chemical Society Reviews, 2016, 45(13): 3731-3746. DOI:10.1039/C5CS00375J |
| [10] | Tsigaridis K, Kanakidou M. Global modelling of secondary organic aerosol in the troposphere:a sensitivity analysis[J]. Atmospheric Chemistry and Physics, 2003, 3(5): 1849-1869. DOI:10.5194/acp-3-1849-2003 |
| [11] | Guo S, Hu M, Guo Q F, et al. Primary sources and secondary formation of organic aerosols in Beijing, China[J]. Environmental Science & Technology, 2012, 46(18): 9846-9853. |
| [12] | Li Y J, Sun Y L, Zhang Q, et al. Real-time chemical characterization of atmospheric particulate matter in China:a review[J]. Atmospheric Environment, 2017, 158: 270-304. DOI:10.1016/j.atmosenv.2017.02.027 |
| [13] | Feng J L, Li M, Zhang P, et al. Investigation of the sources and seasonal variations of secondary organic aerosols in PM2.5 in Shanghai with organic tracers[J]. Atmospheric Environment, 2013, 79: 614-622. DOI:10.1016/j.atmosenv.2013.07.022 |
| [14] | Zhou M, Qiao L P, Zhu S H, et al. Chemical characteristics of fine particles and their impact on visibility impairment in Shanghai based on a 1-year period observation[J]. Journal of Environmental Sciences, 2016, 48: 151-160. DOI:10.1016/j.jes.2016.01.022 |
| [15] | Wang H L, Chen C H, Wang Q, et al. Chemical loss of volatile organic compounds and its impact on the source analysis through a two-year continuous measurement[J]. Atmospheric Environment, 2013, 80: 488-498. DOI:10.1016/j.atmosenv.2013.08.040 |
| [16] |
宫照恒, 薛莲, 孙天乐, 等. 基于高分辨质谱在线观测的2011深圳大运会前后PM1化学组成与粒径分布[J]. 中国科学:化学, 2013, 43(3): 363-372. Gong Z H, Xue L, Sun T L, et al. On-line measurement of PM1 chemical composition and size distribution using a high-resolution aerosol mass spectrometer during 2011 Shenzhen Universiade[J]. Scientia Sinica Chimica, 2013, 43(3): 363-372. |
| [17] | Huang X F, He L Y, Xue L, et al. Highly time-resolved chemical characterization of atmospheric fine particles during 2010 Shanghai World Expo[J]. Atmospheric Chemistry and Physics, 2012, 12(11): 4897-4907. DOI:10.5194/acp-12-4897-2012 |
| [18] | Hu W W, Hu M, Yuan B, et al. Insights on organic aerosol aging and the influence of coal combustion at a regional receptor site of Central Eastern China[J]. Atmospheric Chemistry and Physics, 2013, 13(19): 10095-10112. DOI:10.5194/acp-13-10095-2013 |
| [19] | Ulbrich I M, Canagaratna M R, Zhang Q, et al. Interpretation of organic components from Positive Matrix Factorization of aerosol mass spectrometric data[J]. Atmospheric Chemistry and Physics, 2009, 9(9): 2891-2918. DOI:10.5194/acp-9-2891-2009 |
| [20] | Zhang Q, Jimenez J L, Canagaratna M R, et al. Understanding atmospheric organic aerosols via factor analysis of aerosol mass spectrometry:a review[J]. Analytical and Bioanalytical Chemistry, 2011, 401(10): 3045-3067. DOI:10.1007/s00216-011-5355-y |
| [21] | Middlebrook A M, Bahreini R, Jimenez J L, et al. Evaluation of composition-dependent collection efficiencies for the aerodyne aerosol mass spectrometer using field data[J]. Aerosol Science and Technology, 2012, 46(3): 258-271. DOI:10.1080/02786826.2011.620041 |
| [22] | Huang X F, Xue L, Tian X D, et al. Highly time-resolved carbonaceous aerosol characterization in Yangtze River Delta of China:Composition, mixing state and secondary formation[J]. Atmospheric Environment, 2013, 64: 200-207. DOI:10.1016/j.atmosenv.2012.09.059 |
| [23] | Wang Q Z, Dong X Y, Fu J S, et al. Environmentally dependent dust chemistry of a super Asian dust storm in March 2010:observation and simulation[J]. Atmospheric Chemistry and Physics, 2018, 18(5): 3505-3521. DOI:10.5194/acp-18-3505-2018 |
| [24] |
何瑶, 黄汝锦, 王启元, 等. 我国PM1浓度、化学组分及来源的时空分布[J]. 中国粉体技术, 2017, 23(3): 1-10. He Y, Huang W J, Wang Q Y, et al. Temporal and spatial distribution of atmospheric PM1 concentration, composition and source in China[J]. China Powder Science and Technology, 2017, 23(3): 1-10. |
| [25] | Behera S N, Sharma M. Investigating the potential role of ammonia in ion chemistry of fine particulate matter formation for an urban environment[J]. Science of the Total Environment, 2010, 408(17): 3569-3575. DOI:10.1016/j.scitotenv.2010.04.017 |
| [26] | Khoder M I. Atmospheric conversion of sulfur dioxide to particulate sulfate and nitrogen dioxide to particulate nitrate and gaseous nitric acid in an urban area[J]. Chemosphere, 2002, 49(6): 675-684. DOI:10.1016/S0045-6535(02)00391-0 |
| [27] | Ohta S, Okita T. A chemical characterization of atmospheric aerosol in Sapporo[J]. Atmospheric Environment. Part A. General Topics, 1990, 24(4): 815-822. DOI:10.1016/0960-1686(90)90282-R |
| [28] | Truex T J, Pierson W R, McKee D E. Sulfate in diesel exhaust[J]. Environmental Science & Technology, 1980, 14(9): 1118-1121. |
| [29] | Zhang X Y, Wang Y Q, Zhang X C, et al. Carbonaceous aerosol composition over various regions of China during 2006[J]. Journal of Geophysical Research, 2008, 113(D14): D14111. DOI:10.1029/2007JD009525 |
| [30] | Quan J N, Zhang X S, Zhang Q, et al. Importance of sulfate emission to sulfur deposition at urban and rural sites in China[J]. Atmospheric Research, 2008, 89(3): 283-288. DOI:10.1016/j.atmosres.2008.02.015 |
| [31] | Clegg S L, Brimblecombe P, Wexler A S. Thermodynamic model of the system H+NH4+-SO4+-NO3--H2O at tropospheric temperatures[J]. The Journal of Physical Chemistry A, 1998, 102(12): 2137-2154. DOI:10.1021/jp973042r |
| [32] | Ravishankara A R. Heterogeneous and multiphase chemistry in the troposphere[J]. Science, 1997, 276(5315): 1058-1065. DOI:10.1126/science.276.5315.1058 |
| [33] | Ng N L, Canagaratna M R, Jimenez J L, et al. Real-time methods for estimating organic component mass concentrations from aerosol mass spectrometer data[J]. Environmental Science & Technology, 2011, 45(3): 910-916. |
| [34] | Allan J D, Williams P I, Morgan W T, et al. Contributions from transport, solid fuel burning and cooking to primary organic aerosols in two UK cities[J]. Atmospheric Chemistry and Physics, 2010, 10(2): 647-668. DOI:10.5194/acp-10-647-2010 |
| [35] | Hu W W, Hu M, Hu W, et al. Chemical composition, sources, and aging process of submicron aerosols in Beijing:contrast between summer and winter[J]. Journal of Geophysical Research, 2016, 121(4): 1955-1977. |
| [36] | Qin Y M, Li Y J, Wang H, et al. Particulate matter (PM) episodes at a suburban site in Hong Kong:evolution of PM characteristics and role of photochemistry in secondary aerosol formation[J]. Atmospheric Chemistry and Physics, 2016, 16(22): 14131-14145. DOI:10.5194/acp-16-14131-2016 |
| [37] | Pusede S E, Duffey K C, Shusterman A A, et al. On the effectiveness of nitrogen oxide reductions as a control over ammonium nitrate aerosol[J]. Atmospheric Chemistry and Physics, 2016, 16(4): 2575-2596. DOI:10.5194/acp-16-2575-2016 |
| [38] | Young D E, Kim H, Parworth C, et al. Influences of emission sources and meteorology on aerosol chemistry in a polluted urban environment:results from DISCOVER-AQ California[J]. Atmospheric Chemistry and Physics, 2016, 16(8): 5427-5451. DOI:10.5194/acp-16-5427-2016 |
| [39] | Lin P, Hu M, Deng Z, et al. Seasonal and diurnal variations of organic carbon in PM2.5 in Beijing and the estimation of secondary organic carbon[J]. Journal of Geophysical Research, 2009, 114(D2): D00G11. DOI:10.1029/2008JD010902 |
| [40] | Wang Y, Zhuang G S, Zhang X Y, et al. The ion chemistry, seasonal cycle, and sources of PM2.5 and TSP aerosol in Shanghai[J]. Atmospheric Environment, 2006, 40(16): 2935-2952. DOI:10.1016/j.atmosenv.2005.12.051 |