 2019, Vol. 40
2019, Vol. 40



二次有机气溶胶(secondary organic aerosols, SOA)对我国重霾污染具有重要贡献[1~3], 弄清大气中二次有机气溶胶的主要气体前体物及其贡献, 对有效控制灰霾污染起到至关重要的作用[4].由于形成SOA气体前体物的多样性及其发生的化学与物理过程的复杂性, 以及当前对SOA的分析和观测手段的局限性, 追溯二次有机气溶胶的来源具有很大的挑战性和不确定性. Kleindienst等[5]建立的示踪物产率法(tracer-based method)由于可以估算出特定的二次源对大气颗粒物中有机组分的贡献已经被广泛应用.而示踪物产率法的基本假设之一是示踪物具有良好的稳定性, 但这些示踪物都是饱和及不饱和的烃类物质, 都可能与大气中的氧化剂(·OH、O3和NOx等[6~13])发生反应.虽然针对颗粒物中二次有机示踪物的非均相氧化反应的研究尚未见文献报道, 但是近几年已有学者对一次有机示踪物的非均相氧化反应进行了研究. Knopf等[8]研究了臭氧在生物燃烧排放的一次有机示踪物左旋葡聚糖、脱氢松香酸上非均相氧化反应, 得到摄取系数为1.0×10-5~8.0×10-5, 表明这些一次有机示踪物存在一定的臭氧氧化. Weitkamp等[14]发现烹饪肉类所排放的一次有机示踪物胆固醇、油酸和棕榈烯酸在夏季寿命大约为1d, 并指出以示踪物化学稳定为假设的受体模型在一次源解析上会造成很大的偏差. Dreyfus等[15]在气溶胶袋式反应器中模拟了生物质燃烧的一次有机示踪物胆固醇气溶胶在拟一级条件下与臭氧反应, 实验结果表明:臭氧在胆固醇颗粒上的反应摄取系数为(2.8±0.4)×10-6, 并指出胆固醇虽然是环境有机气溶胶源解析中合适的局部源示踪剂, 但其在大气中的臭氧氧化确实存在.相对而言, 二次有机示踪物本身就是在气体前体物经过氧化之后得到的中间产物, 它们随后所经历的非均相氧化过程可能更加复杂多样, 引起的示踪物产率法预测结果的不确定性可能会更显著.
SOA前体物包括天然源和人为源挥发性有机化合物(volatile organic compounds, VOCs), 其中异戊二烯是排放量最大的非甲烷烃天然源, 全国森林生态系统的排放量为0.03~8.60 Tg·a-1[16, 17], 全球排放量可达到600.00 Tg·a-1[18]; 人为源主要以机动车尾气以及工业排放的芳香族化合物为主, 其中甲苯是人为源SOA前体物的代表, 也是深圳市大气VOCs组成排名第一的物质, 约占总VOCs的22.7%[19].目前多数研究采用2-甲基赤藓糖醇(2-methyl erythritol, 2-ME)[20~29]和2, 3-二羟基-4-氧代戊酸(2, 3-dihydroxy-4-oxopentanoic acid, DHOPA)[22~29]作为二次有机示踪物分别估算异戊二烯和甲苯对SOA的贡献.
本文采用相对速率法[30~35]通过烟雾箱实验测定了不同条件下2-甲基赤藓糖醇的类似物赤藓糖醇(analogue of 2-methyl erythritol, AME)和2, 3-二羟基-4-氧代戊酸(2, 3-dihydroxy-4-oxopentanoic acid, DHOPA)臭氧非均相氧化的有效速率常数, 并结合示踪物产率法分析了两种示踪物氧化对二次源解析的影响.
1 材料与方法 1.1 实验仪器和试剂2 m3室内烟雾箱、恒流雾化气溶胶发生器(美国TSI, 3076)、气相色谱仪(福立9670Ⅱ)、气质联用仪(安捷伦7890A-5975C)、温湿度仪(台湾群特CENTER311)、旋转蒸发仪(东京理化CCA-112, N-1300, OSB-2200)、氮吹仪(东京理化MG-2200)、空气干燥器(无锡汉英GXW2-0.2/1)、空气压缩机(上海捷豹OLF-2530)、微量分析天平(德国赛多利斯MSE3.6P-OCE-DM)、臭氧发生器(Jelight, MODEL600)、臭氧检测仪(2BTechnologies, 106L)、特氟龙滤膜(Pall, Φ 47 mm); 正戊烯(98.0%)、正己烷(≥99.5%)、正二十六烷(99.0%)、2-丁醇(≥99.0%)、赤藓糖醇(≥99.0%)、乙腈(≥99.9%)、D50-二十四烷(98 atom% D)、2, 3-二羟基-4-氧代戊酸(≥98.0%).
1.2 实验方法 1.2.1 烟雾箱实验流程实验开始前, 使用清洁的空气清洗烟雾箱.清洗完成后, 向烟雾箱中通入清洁空气至烟雾箱将要充满的状态.此时通过气溶胶发生器向烟雾箱中通入AME(或DHOPA)和二十六烷气溶胶; 再依次取一定体积的正戊烯和正己烷液体通过玻璃三通进样器随清洁空气挥发进入烟雾箱, 最后利用臭氧发生仪生成臭氧通入烟雾箱.为了消除戊烯和臭氧反应产生的OH对目标物的氧化, 在烟雾箱实验中加入400 μL的2-丁醇作为OH的淬灭剂.每隔1、2、3和6 h检测烟雾箱中气相物质的浓度并用特氟龙滤膜采集250 L体积的气溶胶样品.
1.2.2 气相物质分析实验中每隔特定的时间, 用气相色谱(FID)检测烟雾箱中气相组分的浓度.检测条件:毛细管柱DB-5MS(30 m×0.25 mm×0.25 μm), 柱温50℃, 进样器温度80℃, 检测器温度150℃.
1.2.3 气溶胶样品分析① 预处理:在25 mL的比色管中用1:1的二氯甲烷和乙腈混合液浸润采集气溶胶样品的特氟龙膜, 超声萃取20 min, 静置分层, 上层是溶于乙腈的气溶胶组分样品(AME或DHOPA), 下层是溶于二氯甲烷的气溶胶组分样品(D50-二十四烷和正二十六烷), 分别将上、下两层样品在40℃水浴上用氮吹仪浓缩至300 μL, 低温5℃保存; ②检测分析:在反应瓶中依次加入溶于乙腈的样品200 μL、75 μL BSTFA(含1%的TMC)衍生剂、15 μL吡啶和10 μL 1 mg·L-1的D50-二十四烷内标物, 在60℃恒温水浴锅中反应2 h, 然后用GC-MS进行分析.溶于二氯甲烷的样品经浓缩之后直接注入GC-MS进行分析.检测条件:毛细管柱DB-5MS(30 m×0.25 mm×0.25 μm), 载气为氮气, 流速为1.0 mL·min-1, 进样量为1 μL, 进样口温度250℃, 质谱在电子能量为70eV下以电离源(EI)模式运行, 四极杆温度150℃, 离子源温度230℃.柱箱梯度升温:初始温度80℃, 保持3 min, 然后以5 ℃·min-1的速率升至180℃, 保持2 min, 之后再以10℃·min-1的速率升至300℃, 最后在300℃下运行3 min.根据文献[35, 36]的报道, 利用GC-MS测定AME和DHOPA衍生物时, 选择监测的离子碎片质荷比(m/z)分别为189、205、278、205、395和247、259、275、303、349.
2 相对速率常数分析法Donahue等[37]建立了相对速率常数法用来分析烟雾箱实验中的气相和气溶胶相数据以获得某种目标物的有效速率常数.在研究气溶胶组分的非均相氧化时, 采用相对速率常数法, 通过比较同一氧化体系中某一气相物质(其氧化反应速率常数已知)和气溶胶中待测组分的浓度变化趋势, 分析计算得到气溶胶中该物质的有效速率常数.关于相对速率法的基本理论及公式推导可具体参考文献[35, 36].在进行相对速率测试的烟雾箱实验时, 为了减少烟雾箱壁损失引起的实验误差, 选取不参与反应的正二十六烷和正己烷作为内标物分别对AME或DHOPA和戊烯的测定浓度进行校正.气溶胶待测物质i(AME或DHOPA)相对于气相参考物r(戊烯)臭氧氧化的反应速率的有效比值和有效速率常数可以表达成公式(1)和公式(2).基于戊烯和正己烷的浓度比值与AME或DHOPA和正二十六烷的浓度比值的相关关系来确定示踪物的反应速率.
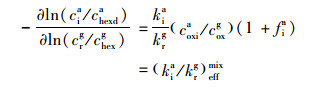
|
(1) |
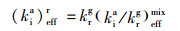
|
(2) |
式中, a和g分别表示气溶胶和气相; i表示气溶胶中的目标组分(如AME或DHOPA); r表示气相中的参考物(如戊烯); hexd和hex分别表示正二十六烷和正己烷; chexda和chexg分别表示正二十六烷和正己烷的分数(10-9); coxia和coxg分别表示气溶胶中臭氧的平均浓度和气相臭氧体积浓度(10-9); kia和krg分别表示气溶胶组分i和气相参考物r的反应速率常数; (kia/krg)effmix表示混合相中气溶胶组分i和气相参考物r的反应速率常数的有效比; (kia)effr表示气溶胶组分i相对气相参考物r的有效速率常数.
3 结果与讨论 3.1 气溶胶中二次示踪物的相对速率常数根据烟雾箱实验测得的AME或DHOPA、戊烯以及内标物的浓度, 采用相对速率法分析获得不同环境条件(初始臭氧的体积浓度和相对湿度)和不同气溶胶混合形式及混合比例条件下, AME或DHOPA臭氧非均相氧化的有效速率常数, 如表 1所示.
|
|
表 1 不同条件下AME和DHOPA的有效速率常数1) Table 1 Effective rate constants of AME and DHOPA under different conditions |
以烟雾箱中臭氧体积分数为150.0×10-9、RH=70.0%的室温(25.5℃±1.0℃)条件下进行的AME和DHOPA臭氧非均相氧化的实验为例. 图 1显示的是在臭氧非均相氧化实验进行0、1、2、3、6 h时, AME或DHOPA与正二十六烷的浓度比(concentration ratio, CR)的对数值[ln (AME或DHOPA/正二十六烷, CR)]和戊烯与正己烷的浓度比的对数值[ln (戊烯/正己烷, CR)]的相关关系, 所拟合的曲线方程分别为y=0.054 7x+0.000 1(AME)和y=0.074 7x-0.085 6(DHOPA).由式(1)可知, 拟合曲线的斜率即AME和DHOPA相对戊烯臭氧氧化反应的速率常数的有效比值, 分别为0.054 7和0.074 7.该AME和DHOPA臭氧非均相氧化实验中, 实测的戊烯臭氧反应速率常数分别为9.49×10-18cm3·(molecule·s)-1和9.23×10-18cm3·(molecule·s)-1.综上分析得出AME和DHOPA气溶胶臭氧非均相氧化的有效速率常数分别为5.19×10-19 cm3·(molecule·s)-1和6.89×10-19 cm3·(molecule·s)-1.根据相同条件下重复的烟雾箱实验数据以及气相和气溶胶相样品中待测组分和标准物含量的平行样测定结果, 得出AME或DHOPA气溶胶臭氧非均相氧化的有效速率常数的相对标准偏差分别小于7.21%和5.68%.

|
图 1 混合相AME或DHOPA浓度比值与戊烯浓度比值的相关关系 Fig. 1 Mixed phase concentration ratio-ratio plot of AME and DHOPA compared to pentene |
图 2显示的是上述两组烟雾箱实验中t时刻气溶胶AME(或DHOPA)与正二十六烷浓度比(CRt)与其初始浓度比(CR0)的比值[AME(或DHOPA)/正二十六烷: CRt/CR0], 以及反应过程中t时刻正二十六烷浓度(C26烷, t)与其初始浓度(C26烷, 0)的比值(正二十六烷: C26烷, t/C26烷, 0), 随时间的变化.结果表明前3 h内, AME和DHOPA分别被氧化20.0%和25.0%, 后3 h中, AME和DHOPA的氧化趋于缓慢, 均不超过5.0%;在整个烟雾箱实验过程中, 正二十六烷浓度与其初始浓度比的数据点基本分布在比值为0.953的直线上, 说明正二十六烷气溶胶在实验过程中并不发生反应.

|
图 2 烟雾箱实验过程中各物质浓度比值随时间的变化 Fig. 2 Changes in concentration ratios over time in the course of the chamber experiments |
表 1列出了本研究所有烟雾箱实验所测得的AME和DHOPA的有效速率常数.通过比较相同的条件下(初始臭氧体积分数和相对湿度等)DHOPA和AME非均相氧化的有效速率常数, 发现平均来看, DHOPA气溶胶臭氧非均相氧化的反应速率较AME气溶胶快32.1%.
AME和DHOPA都是饱和结构, AME是内消旋的丁四醇, 端链醇能够进一步氧化成酸, 而DHOPA是不含端链醇的α-碳上连羰基的戊酸.一般来说, 醇羟基活泼氢的化学位移在0.5×10-6~5.5×10-6之间, 从氢的化学位移上看(如图 3), AME的还原性强于DHOPA, 更易被氧化.综合该结构分析, AME气溶胶臭氧氧化的速率应该略高于DHOPA, 而实验结果表明DHOPA的氧化速率要高于AME, 这说明直接与臭氧的反应, 醇羟基的反应活性并不占优势, 而DHOPA中的α-碳上的羰基比AME上的醇羟基更容易被臭氧氧化, 导致DHOPA气溶胶臭氧氧化的有效速率常数是AME气溶胶臭氧氧化的有效速率常数的1.32倍左右.

|
图 3 AME和DHOPA的氢化学位移[38] Fig. 3 Hydrogen chemical shift of AME and DHOPA |
基于深圳市实际臭氧体积分数水平, 本研究的初始臭氧体积分数范围在100.0×10-9~200.0 ×10-9, 并依据该范围划分3个水平:100.0×10-9、150.0×10-9、200.0×10-9.
图 4显示的是在不同初始臭氧体积分数下, 在臭氧非均相氧化过程中, t时刻AME或DHOPA浓度与正二十六烷浓度比(CRt)与其初始浓度比(CR0)的比值(AME或DHOPA/正二十六烷: CRt/CR0)随时间的变化.结果表明在6 h的烟雾箱实验中, 初始臭氧体积分数为104.1×10-9、148.5×10-9和205.8 ×10-9时, AME分别被臭氧氧化了5.0%、7.0%和10.0%, 且AME臭氧非均相氧化是一个先快后慢的过程, 在0~3h和3~6 h的氧化过程中, 分别被氧化了8.0%和4.0%.在6 h的DHOPA臭氧非均相氧化的烟雾箱实验中, 初始臭氧体积分数为96.7×10-9、164.5×10-9和200.6×10-9时, DHOPA分别被臭氧氧化了14.0%、16.5%和18.0%, 0~3 h内DHOPA的氧化量是3~6 h中DHOPA的3~5倍, 表明DHOPA臭氧氧化的速度逐渐降低.这可能是因为随着反应时间的增加, DHOPA的浓度逐渐减少, 与臭氧分子的碰撞频率降低, 导致反应速度趋于缓慢.总体来说:随着初始臭氧体积分数的升高, 被臭氧氧化的AME和DHOPA逐渐增加.而在深圳大气环境中, 夏季的日平均臭氧体积分数(78.2×10-9)高于冬季(34.5×10-9)[39], 依此实验结果, 可以推测深圳夏季大气环境中的二次示踪物2-ME和DHOPA气溶胶的氧化要比冬季更快.

|
图 4 不同初始臭氧体积分数下AME和DHOPA的氧化 Fig. 4 Oxidation of AME and DHOPA with ozone under different initial ozone concentrations |
本研究分别在RH=70%和RH=90%的水平下考察了相对湿度对AME和DHOPA臭氧非均相氧化的影响.结果表明在6h烟雾箱实验中, 在RH=70.2%和RH=89.6%的条件下, AME分别被臭氧氧化了9.8%和9.6%, 如图 5(a)所示; 在RH=89.8%和RH=72.3%的条件下, DHOPA分别被臭氧氧化了16.4%和18.9%, 如图 5(b)所示.

|
图 5 不同相对湿度下AME和DHOPA的氧化 Fig. 5 Oxidation of AME and DHOPA by ozone under different relative humidity |
AME和DHOPA臭氧非均相氧化的有效速率常数均在相对较高湿度(RH=90%)的条件下略低, 这可能是因为高湿度条件下, 气溶胶表面会形成水膜增加了臭氧的传质阻力, 降低了反应活性. Han等[40]发现随着相对湿度的升高, NO2在煤烟气溶胶表面的摄取系数逐渐降低, 认为在较高相对湿度的条件下, 煤烟的吸水性会导致其颗粒物的孔隙变得更加致密, 抑制反应气体NO2在煤烟颗粒物孔隙中的扩散, 导致非均相氧化反应速率减慢.
3.4 混合状态的影响本研究中的混合状态指不同AME和DHOPA气溶胶的混合比例及混合形式对AME或DHOPA气溶胶臭氧非均相氧化反应的影响, 考察的气溶胶混合形式分为“外混”和“内混”:“外混”是指AME和DHOPA的气溶胶发生液分别注入恒流雾化气溶胶发生器中, 产生的气溶胶先后注入烟雾箱再混合的形式; “内混”是指AME和DHOPA的混合溶液作为气溶胶发生液由恒流雾化气溶胶发生器一起注入烟雾箱的混合形式.
图 6展示的是不同外混比例下, 臭氧氧化过程中DHOPA与正二十六烷的浓度比(CRt)与其初始浓度比(CR0)的比值的对数[ln(DHOPA/正二十六烷, CRt/CR0)]和AME与正二十六烷的浓度比(CRt)与其初始浓度比(CR0)的比值的对数[ln(AME/正二十六烷, CRt/CR0)], 所拟合的直线斜率表明了非均相氧化过程中DHOPA和AME反应速率的倍数差异.当AME:DHOPA为5:1、1:1和1:5时, DHOPA气溶胶臭氧非均相氧化的反应速率分别是AME反应速率的1.32、1.46和1.78倍.结果表明:在本实验条件范围内, 随着DHOPA气溶胶在外混气溶胶中比例的增加, DHOPA气溶胶臭氧非均相氧化的反应速率较AME气溶胶臭氧非均相氧化的反应速率越来越快.

|
图 6 不同外混比例下AME和DHOPA气溶胶臭氧氧化反应速率的比较 Fig. 6 Comparisons of the ozone oxidation rates of AME and DHOPA among various external mixing compositions |
图 7展示的是内混体系中不同混合比例AME和DHOPA气溶胶臭氧非均相氧化过程中, t时刻AME或DHOPA浓度与正二十六烷浓度比(CRt)与其初始浓度比(CR0)的比值(AME或DHOPA/正二十六烷: CRt/CR0)随时间的变化.

|
图 7 不同内混比例对AME和DHOPA氧化的影响 Fig. 7 Influence of internal mixing compositions to the ozone oxidation of AME and DHOPA |
结果表明在6 h的烟雾箱实验中, 随着内混气溶胶体系中DHOPA占比越高, AME被臭氧氧化的越少, 而DHOPA的氧化速率越快, 与AME:DHOPA=5:1相比, AME:DHOPA=1:1时, AME少氧化3.0%而DHOPA多氧化5.0%, 由此推断:在混合气溶胶中占比较高的物质被臭氧氧化的速率越大.本实验结果还表明, 内外混合条件下, AME或DHOPA气溶胶臭氧非均相氧化的有效速率常数分别为4.38×10-19 cm3·(molecule·s)-1、4.45×10-19 cm3·(molecule·s)-1和6.55×10-19 cm3·(molecule·s)-1、6.50×10-19 cm3·(molecule·s)-1.因此, 本实验条件中气溶胶的外混和内混对AME和DHOPA气溶胶臭氧非均相氧化的有效速率常数并无显著影响.
3.5 示踪物产率法的不确定性分析 3.5.1 示踪物产率法示踪物产率法是指通过烟雾箱实验模拟特定VOC生成SOA, 测定该VOC反应生成的SOA中二次有机示踪物的浓度, 从而确定示踪物在SOA中的质量分数, 利用该质量分数, 结合测定的大气气溶胶中各前体物所对应的示踪物浓度, 推算源于该VOC的SOA对大气有机气溶胶的贡献, 如公式(3)和公式(4)所示[5].
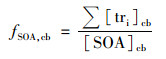
|
(3) |
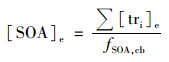
|
(4) |
式中, Σ[tri]cb为烟雾箱中特定前体物生成SOA中示踪物浓度之和(μg·m-3); [SOA]cb为烟雾箱中特定前体物生成的SOA浓度(μg·m-3); fSOA, cb为烟雾箱中特定前体物SOA示踪物浓度和占总SOA的质量分数; Σ[tri]e为大气中特定前体物生成SOA中的示踪物浓度之和(μg·m-3); [SOA]e为大气中特定前体物生成的SOA浓度(μg·m-3).
3.5.2 气溶胶的大气有效寿命二次有机示踪物的臭氧非均相氧化反应是一个二级反应, 其反应速度与两反应物浓度的乘积成正比, 也就是与反应物浓度的二次方成正比.二级反应的反应速率方程式如式(5), 当大气臭氧体积分数保持不变时.方程式可写成拟一级反应方程如式(6), 对式(6)进行积分变换得式(7)[7].
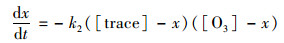
|
(5) |
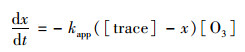
|
(6) |
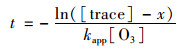
|
(7) |
式中, t为时间(s); [O3]为臭氧体积浓度(molecule·cm-3); x为示踪物的变化浓度(molecule·cm-3); [tracer]为二次示踪物浓度(molecule·cm-3); k2为二级反应速率常数[cm3·(molecule·s)-1]; kapp为准一级速率常数, 也常被称拟一级速率常数[cm3·(molecule·s)-1].
在计算气溶胶的大气有效寿命时一般按气溶胶反应物衰减到原浓度的1/e作为计算节点, 基于公式(7), 根据二次示踪物的变化浓度以及测得的有效速率常数(即式中准一级速率常数)得到的大气有效寿命也称e倍寿命.由于在不同环境条件下, AME和DHOPA臭氧非均相氧化的有效速率常数不同, 使得它们的大气有效寿命也有所不同.为了明确在估算异戊二烯和甲苯对SOA贡献时, AME和DHOPA的臭氧非均相氧化可能引起的不确定性范围, 本研究基于大气臭氧体积分数为75×10-9的假设和测得的AME(2-ME类似物)和DHOPA气溶胶臭氧非均相氧化有效速率常数的极值(极大值:RH=70.0%条件下获得; 极小值:混合气溶胶体系中各自占比最低的条件下获得), 计算得到不同条件下所有烟雾箱实验中AME和DHOPA臭氧非均相氧化的有效速率常数和有效寿命的极大值和极小值, 如表 2所示.结果表明, 当只考虑臭氧对AME和DHOPA的非均相氧化时, AME和DHOPA的大气有效寿命分别是11.0~14.6 d和8.1~9.4 d.
|
|
表 2 AME和DHOPA有效速率常数和有效寿命的极值 Table 2 Boundary values of effective rate constant and effective life of AME and DHOPA |
3.5.3 二次有机示踪物的衰减模拟
基于AME(2-ME的类似物)和DHOPA气溶胶臭氧非均相氧化过程, 模拟了30 d内大气颗粒物中2-ME和DHOPA的衰减, 如图 8和图 9所示.结果表明夏季气溶胶暴露于体积分数75 ×10-9臭氧5 d后, 大约32.4%~44.8%的2-ME和40.5%~47.3%的DHOPA会被氧化, 这使得以2-ME和DHOPA为示踪物的示踪物产率法将会低估异戊二烯和甲苯对SOA的贡献约32.4%~44.8%和40.5%~47.3%;冬季气溶胶暴露于体积分数35×10-9臭氧5 d后, 大约16.5%~21.3%的2-ME和18.3%~24.8%的DHOPA会被氧化, 这使得应用2-ME和DHOPA为示踪物的示踪物产率法将会低估异戊二烯和甲苯对SOA的贡献, 约16.5%~21.3%和18.3%~24.8%.

|
图 8 夏季和冬季2-ME臭氧非均相氧化过程中的衰减模拟 Fig. 8 Decay simulation of ozone oxidation of 2-ME in summer and winter |

|
图 9 夏季和冬季DHOPA臭氧非均相氧化过程中的衰减模拟 Fig. 9 Decay simulation of ozone oxidation of DHOPA in summer and winter |
2-ME和DHOPA气溶胶臭氧非均相氧化过程中的衰减模拟结果表明, 臭氧非均相氧化对示踪物产率法解析二次源贡献时会造成较大偏差, 但本研究通过实验获得的这种偏差还不能够完全用于校正示踪物产率法对二次源贡献的估算结果.首先, 在大尺度范围的环境条件下, 这两种气溶胶粒子的大气停留时间尚不清楚; 其次, 这种非均相氧化与气溶胶组成等条件有关, 不同环境来源的气溶胶粒子可能有不同的非均相氧化速率; 另外, 本研究没有考虑OH自由基对二次有机示踪物的非均相氧化过程对示踪物产率法估算准确性的影响.虽然二次有机示踪物的氧化反应对示踪物产率法的准确性提出了质疑, 但示踪物的特异性使示踪物产率法在未来的源解析过程中仍被广泛使用, 所以今后的相关研究应该考虑二次有机示踪物氧化的影响.本文是对二次有机示踪物非均相氧化的初步探索, 其理论研究(如示踪物的氧化反应机制等)及实际应用(如对示踪物产率法准确性的校正等)还有待于更深入地探讨.
4 结论(1) 初始臭氧体积浓度对AME和DHOPA气溶胶的臭氧非均相氧化反应有显著影响, 臭氧体积浓度越高, 气溶胶被氧化得更多. AME和DHOPA臭氧非均相氧化的有效速率常数均在较高相对湿度(RH=90.0%)的条件下略低.
(2) 在本实验条件范围内, 内混实验结果与外混实验结果类似, 随着DHOPA气溶胶在外混或内混气溶胶中比例的增加, DHOPA气溶胶臭氧非均相氧化的反应速率较AME气溶胶臭氧非均相氧化的反应速率越来越快.
(3) 通过烟雾箱实验测得的AME和DHOPA臭氧非均相氧化速率分别为(4.60±0.66)×10-19 cm3·(molecule·s)-1和(6.57±0.51)×10-19 cm3·(molecule·s)-1, 仅针对O3的氧化过程, AME和DHOPA大气寿命分别为11.0~14.6 d和8.1~9.4 d, 若2-ME和DHOPA两种二次有机示踪物暴露于深圳夏季大气环境中5 d, 异戊二烯和甲苯对SOA贡献的结果分别低估32.4%~44.8%和40.5%~47.3%, 若暴露于深圳冬季大气环境中5 d异戊二烯和甲苯对SOA贡献的结果分别低估16.5%~21.3%和18.3%~24.8%.
| [1] | Chen R J, Zhao H Z, Kan H D. Heavy smog and hospital visits in Beijing, China[J]. American Journal of Respiratory and Critical Care Medicine, 2013, 188(9): 1170-1171. DOI:10.1164/rccm.201304-0678LE |
| [2] | Huang R J, Zhang Y L, Bozzetti C, et al. High secondary aerosol contribution to particulate pollution during haze events in China[J]. Nature, 2014, 514(7521): 218-222. DOI:10.1038/nature13774 |
| [3] |
陈多宏, 张涛, 蒋斌, 等. 典型二次有机气溶胶危害性及环境监测技术研究进展[J]. 环境科学与技术, 2014, 37(S2): 305-309. Chen D H, Zhang T, Jiang B, et al. Hazards and environmental monitoring technology progress for typical secondary organic aerosols is reviewed[J]. Environmental Science & Technology, 2014, 37(S2): 305-309. |
| [4] |
郭松, 胡敏, 郭庆丰, 等. 二次有机气溶胶估算方法比较研究[J]. 化学学报, 2014, 72(6): 658-666. Guo S, Hu M, Guo Q F, et al. Comparison of secondary organic aerosol estimation methods[J]. Acta Chimica Sinica, 2014, 72(6): 658-666. |
| [5] | Kleindienst T E, Jaoui M, Lewandowski M, et al. Estimates of the contributions of biogenic and anthropogenic hydrocarbons to secondary organic aerosol at a southeastern US location[J]. Atmospheric Environment, 2007, 41(37): 8288-8300. DOI:10.1016/j.atmosenv.2007.06.045 |
| [6] | Kessler S H, Smith J D, Che D L, et al. Chemical sinks of organic aerosol:kinetics and products of the heterogeneous oxidation of erythritol and levoglucosan[J]. Environmental Science & Technology, 2010, 44(18): 7005-7010. |
| [7] | Chapleski Jr R C, Zhang Y F, Troya D, et al. Heterogeneous chemistry and reaction dynamics of the atmospheric oxidants, O3, NO3, and OH, on organic surfaces[J]. Chemical Society Reviews, 2016, 45(13): 3731-3746. DOI:10.1039/C5CS00375J |
| [8] | Knopf D A, Forrester S M, Slade J H. Heterogeneous oxidation kinetics of organic biomass burning aerosol surrogates by O3, NO2, N2O5, and NO3[J]. Physical Chemistry Chemical Physics, 2011, 13(47): 21050-21062. DOI:10.1039/c1cp22478f |
| [9] | Hearn J D, Smith G D. Kinetics and product studies for ozonolysis reactions of organic particles using aerosol CIMS[J]. The Journal of Physical Chemistry A, 2004, 108(45): 10019-10029. DOI:10.1021/jp0404145 |
| [10] | Zeng G, Holladay S, Langlois D, et al. Kinetics of heterogeneous reaction of ozone with linoleic acid and its dependence on temperature, physical state, RH, and ozone concentration[J]. The Journal of Physical Chemistry A, 2013, 117(9): 1963-1974. DOI:10.1021/jp308304n |
| [11] | Weitkamp E A, Lambe A T, Donahue N M, et al. Laboratory measurements of the heterogeneous oxidation of condensed-phase organic molecular makers for motor vehicle exhaust[J]. Environmental Science & Technology, 2008, 42(21): 7950-7956. |
| [12] | Katrib Y, Biskos G, Buseck P R, et al. Ozonolysis of mixed oleic-acid/stearic-acid particles:reaction kinetics and chemical morphology[J]. The Journal of Physical Chemistry A, 2005, 109(48): 10910-10919. DOI:10.1021/jp054714d |
| [13] | Hung H M, Katrib Y, Martin S T. Products and mechanisms of the reaction of oleic acid with ozone and nitrate radical[J]. The Journal of Physical Chemistry A, 2005, 109(20): 4517-4530. DOI:10.1021/jp0500900 |
| [14] | Weitkamp E A, Hartz K E H, Sage A M, et al. Laboratory measurements of the heterogeneous oxidation of condensed-phase organic molecular makers for meat cooking emissions[J]. Environmental Science & Technology, 2008, 42(14): 5177-5182. |
| [15] | Dreyfus M A, Tolocka M P, Dodds S M. Cholesterol ozonolysis:kinetics, mechanism, and oligomer products[J]. The Journal of Physical Chemistry A, 2005, 109(28): 6242-6248. DOI:10.1021/jp050606f |
| [16] |
张莉, 王效科, 欧阳志云, 等. 中国森林生态系统的异戊二烯排放研究[J]. 环境科学, 2003, 24(1): 8-15. Zhang L, Wang X K, Ouyang Z Y, et al. Estimation of isoprene emission from forest ecosystems in China[J]. Environmental Science, 2003, 24(1): 8-15. |
| [17] | Fu Y, Liao H. Biogenic isoprene emissions over China:sensitivity to the CO2 inhibition effect[J]. Atmospheric and Oceanic Science Letters, 2016, 9(4): 277-284. DOI:10.1080/16742834.2016.1187555 |
| [18] | Guenther A, Karl T, Harley P, et al. Estimates of global terrestrial isoprene emissions using MEGAN (Model of emissions of gases and aerosols from nature)[J]. Atmospheric Chemistry and Physics, 2006, 6(11): 3181-3210. DOI:10.5194/acp-6-3181-2006 |
| [19] |
朱少峰, 黄晓锋, 何凌燕, 等. 深圳大气VOCs浓度的变化特征与化学反应活性[J]. 中国环境科学, 2012, 32(12): 2140-2148. Zhu S F, Huang X F, He L Y, et al. Variation characteristics and chemical reactivity of ambient VOCs in Shenzhen[J]. China Environmental Science, 2012, 32(12): 2140-2148. |
| [20] | Fu P Q, Kawamura K, Kanaya Y, et al. Contributions of biogenic volatile organic compounds to the formation of secondary organic aerosols over Mt. Tai, Central East China[J]. Atmospheric Environment, 2010, 44(38): 4817-4826. DOI:10.1016/j.atmosenv.2010.08.040 |
| [21] | Bai J, Sun X M, Zhang C X, et al. The OH-initiated atmospheric reaction mechanism and kinetics for levoglucosan emitted in biomass burning[J]. Chemosphere, 2013, 93(9): 2004-2010. DOI:10.1016/j.chemosphere.2013.07.021 |
| [22] | Offenberg J H, Lewandowski M, Jaoui M, et al. Contributions of biogenic and anthropogenic hydrocarbons to secondary organic aerosol during 2006 in research Triangle Park, NC[J]. Aerosol and Air Quality Research, 2011, 11(2): 99-108. DOI:10.4209/aaqr.2010.11.0102 |
| [23] | Hu D, Bian Q J, Li T W Y, et al. Contributions of isoprene, monoterpenes, β-caryophyllene, and toluene to secondary organic aerosols in Hong Kong during the summer of 2006[J]. Journal of Geophysical Research, 2008, 113(D22): D22206. DOI:10.1029/2008JD010437 |
| [24] | Feng J L, Li M, Zhang P, et al. Investigation of the sources and seasonal variations of secondary organic aerosols in PM2.5 in Shanghai with organic tracers[J]. Atmospheric Environment, 2013, 79: 614-622. DOI:10.1016/j.atmosenv.2013.07.022 |
| [25] | Rutter A P, Snyder D C, Stone E A, et al. Preliminary assessment of the anthropogenic and biogenic contributions to secondary organic aerosols at two industrial cities in the Upper Midwest[J]. Atmospheric Environment, 2014, 84: 307-313. DOI:10.1016/j.atmosenv.2013.11.014 |
| [26] | Fu P Q, Kawamura K, Chen J, et al. Secondary production of organic aerosols from biogenic VOCs over Mt. Fuji, Japan[J]. Environmental Science & Technology, 2014, 48(15): 8491-8497. |
| [27] | Guo S, Hu M, Guo Q F, et al. Primary sources and secondary formation of organic aerosols in Beijing, China[J]. Environmental Science & Technology, 2012, 46(18): 9846-9853. |
| [28] | Kleindienst T E, Jaoui M, Lewandowski M, et al. The formation of SOA and chemical tracer compounds from the photooxidation of naphthalene and its methyl analogs in the presence and absence of nitrogen oxides[J]. Atmospheric Chemistry and Physics, 2012, 12(18): 8711-8726. DOI:10.5194/acp-12-8711-2012 |
| [29] | Ding X, Wang X M, Gao B, et al. Tracer-based estimation of secondary organic carbon in the Pearl River Delta, South China[J]. Journal of Geophysical Research, 2012, 117(D5): D05313. |
| [30] | Medcraft C, Robertson E G, Thompson C D, et al. Infrared spectroscopy of ozone and hydrogen chloride aerosols[J]. Physical Chemistry Chemical Physics, 2009, 11(36): 7848-7852. DOI:10.1039/b905424n |
| [31] | Lelièvre S, Bedjanian Y, Pouvesle N, et al. Heterogeneous reaction of ozone with hydrocarbon flame soot[J]. Physical Chemistry Chemical Physics, 2004, 6(6): 1181-1191. DOI:10.1039/B316895F |
| [32] | Lambe A T, Miracolo M A, Hennigan C J, et al. Effective rate constants and uptake coefficients for the reactions of organic molecular markers (n-alkanes, hopanes, and steranes) in motor oil and diesel primary organic aerosols with hydroxyl radicals[J]. Environmental Science & Technology, 2009, 43(23): 8794-8800. |
| [33] | Dilbeck C W, Finlayson-Pitts B J. Heterogeneous oxidation of a phosphocholine on synthetic sea salt by ozone at room temperature[J]. Physical Chemistry Chemical Physics, 2013, 15(6): 1990-2002. DOI:10.1039/C2CP43665E |
| [34] | Esteve W, Budzinski H, Villenave E. Relative rate constants for the heterogeneous reactions of NO2 and OH radicals with polycyclic aromatic hydrocarbons adsorbed on carbonaceous particles. Part 2:PAHs adsorbed on diesel particulate exhaust SRM 1650a[J]. Atmospheric Environment, 2006, 40(2): 201-211. DOI:10.1016/j.atmosenv.2005.07.053 |
| [35] | Jaoui M, Kleindienst T E, Lewandowski M, et al. Identification and quantification of aerosol polar oxygenated compounds bearing carboxylic or hydroxyl groups. 1. Method development[J]. Analytical Chemistry, 2004, 76(16): 4765-4778. DOI:10.1021/ac049919h |
| [36] | Hartz K E H, Weitkamp E A, Sage A M, et al. Laboratory measurements of the oxidation kinetics of organic aerosol mixtures using a relative rate constants approach[J]. Journal of Geophysical Research, 2007, 112(D4): 204. |
| [37] | Donahue N W, Robinson A L, Hartz K E H, et al. Competitive oxidation in atmospheric aerosols:the case for relative kinetics[J]. Geophysical Research Letters, 2005, 32(16): 247-275. |
| [38] | 秦海林, 于德泉. 天然有机化合物核磁共振氢谱集[M]. 北京: 化学工业出版社, 2011: 59-78. |
| [39] |
廖宇强, 许舒婕. 深圳近地面臭氧来源分析及空气质量评价探讨——以洪湖子站及南湖子站为例[J]. 广东化工, 2014, 41(11): 183-185, 189. Liao Y Q, Xu S J. Sources analysis of ozone near the ground in Shenzhen and assessment of air quality——Take Honghu lake sub-stations and Nanhu lake sub-stations as an example[J]. Guangdong Chemical Industry, 2014, 41(11): 183-185, 189. |
| [40] | Han C, Liu Y C, He H. Heterogeneous reaction of NO2 with soot at different relative humidity[J]. Environmental Science and Pollution Research, 2017, 24(26): 21248-21255. DOI:10.1007/s11356-017-9766-y |