 2019, Vol. 40
2019, Vol. 40



2. 烟台大学土木工程学院, 烟台 264005
2. School of Civil Engineering, Yantai University, Yantai 264005, China
水体砷(As)污染严重威胁人类健康[1].水体砷的污染主要来源于吸附态砷的释放[2].自然界中砷主要是吸附在铁氧化物上, 如针铁矿、水铁矿、赤铁矿等[3~6].铁相溶解和砷形态转变是促进吸附态砷释放至水体的主要因素[7, 8].
微生物在砷的迁移转化过程中起着至关重要的作用, 它们通过改变吸附载体和吸附态砷的赋存形态及价态影响砷的迁移转化[9, 10].近年来, 人们发现硫酸盐还原菌(SRB)介导的硫酸盐还原过程间接参与了砷的生物地球化学循环[11~14].生物产生的负二价硫(S2-), 可将五价砷[As(Ⅴ)]还原为三价砷[As(Ⅲ)], As(Ⅲ)与S2-生成硫化砷(AsS/As2S3)沉淀从而固定砷[15].关于砷的还原过程是原位固相还原还是砷释放之后再还原还存在争议[16~18]. Ohtsuka等[16]的研究发现, 吸附在铁氧化物表面的As(Ⅴ)可被直接还原为As(Ⅲ).相反, Huang等[17]在水溶液中证明As(Ⅴ)先从氧化铁中解吸, 然后被Shewanella putrefaciens菌株CN-32还原成As(Ⅲ). Luo等[18]认为吸附在固体表面的As(Ⅴ)的还原是可以忽略的, 或至少比溶解的As(Ⅴ)在水相中的还原要慢得多.此外, 能否生成硫化砷沉淀与S/As浓度比、pH、Eh密切相关[19~23]. S2-/As浓度比越高越有可能生成硫化砷沉淀, 但S2-若进一步增加, As2S3沉淀可能由于砷-硫络合物的形成而变得可溶[18]; pH值与水中砷含量呈正相关性, 在中性或偏酸性时有利于As(Ⅲ)存在[24]; Eh越高, 砷越容易被吸附, Eh持续下降会导致砷释放增加, 但在强还原条件下能生成硫化砷矿物[21].近年来, 微生物菌群介导下的吸附态砷的迁移转化研究非常多[18, 25~28].而关于单一硫酸盐还原菌介导下砷的迁移转化机制的研究是较少的[18].
另一方面, 硫酸盐还原菌产生的S2-还可还原固相铁矿物至二价铁, 从而形成二次铁矿-硫化铁矿.硫化铁矿可再次吸附释放出来的As[29, 30]. Couture等[31]的研究发现, 硫化铁矿对As(Ⅴ)的吸附能力大于原始铁矿物, 此过程会再次固定溶解态砷; 对于As(Ⅲ), 硫化铁矿的吸附能力不如原始铁矿物.与此同时, 固相铁的还原溶解会促进砷的释放[7, 23].因而, 二次铁矿再次固定砷还是促进砷的释放取决于铁矿物的变化.在SRB作用下, 铁氧化物会如何转变, 这种转变对吸附态砷又会产生怎样的影响, 还有待研究.
本文的目的是结合液相溶解态砷和固相吸附态砷的形态转化, 研究SRB对吸附态砷的迁移转化过程, 以期为自然界中吸附态砷的释放机制提供理论依据.
1 材料与方法 1.1 实验药品氢氧化钾(KOH, 优级纯)、磷酸氢二胺[(NH4)2HPO4, 分析纯]、磷酸氢二钾(K2HPO4, 分析纯)、硝酸铁[Fe(NO3)3·9H2O]、浓盐酸(HCl, 优级纯)、乳酸钠(C3H5O3Na, 化学纯)均由国药集团化学试剂(北京)有限公司生产.硼氢化钾(KBH4, 原子荧光专用)购自天津南开允公合成技术有限公司, 氢氧化钠(GR, 97%, 片状)购自阿拉丁公司, 改良巴尔斯氏培养基基础购自山东拓普生物工程有限公司.
1.2 实验菌株本研究选用的菌种为本实验室分离的菌株Desulfovibrio vulgaris DP4, 能在厌氧条件下以乳酸根作为电子供体, 硫酸根作为电子受体供其生长.
1.3 培养基的制备菌株生长培养基为改良巴尔斯氏培养基, 称取改良巴尔斯氏培养基基础10 g于1 L去离子水(加热煮沸30 min并充氮气直至冷却)中使其完全溶解, 分装后各充10 min氮气再次除氧, 121℃高压灭菌30 min, 冷却至常温.每100 mL添加1支S2211(含0.35 g乳酸钠和0.05 g磷酸氢二钾), 混匀, 备用.
1.4 针铁矿的制备参照Schwertmann等[32]修正的方法, 称取48.5 g的Fe(NO3)3·9H2O, 24.6 g的KOH分别装于200 mL和100 mL的塑料杯中.将KOH溶液逐滴加入至Fe(NO3)3·9H2O中, 并不断搅拌.接着将上述混合液稀释至400 mL.水浴加热70℃, 持续60 h后, 用乙醇清洗两遍.最后在真空干燥箱60℃下干燥2 h即可.
1.5 实验方法称量100 mg的针铁矿于50 mL的离心管中, 加入2 mL配制好的1 μmol·L-1的As(Ⅴ)溶液并用超纯水定容到50 mL, 盖上盖子放置转盘12 h使其吸附.将其取下进行12 000 r·min-1、5 min离心, 取上清液保存, 然后用超纯水清洗3次去除未吸附的砷, 最终针铁矿上的吸附态砷浓度为1.2 g·kg-1, 接近实际高砷污染土壤中的砷浓度[33].最后用封口膜包裹进行灭菌.对保存在-80℃冰箱中的DP4菌种在厌氧箱中进行活化, 在接入2%的DP4保存菌液于100 mL的新鲜培养基中, 然后放置于30℃下恒温培养.所有实验操作均是在厌氧箱(N2环境, 氧气含量低于0.1 mg·L-1)中完成.将100 mg已灭过菌并吸附了砷的针铁矿加入到100 mL培养基中.将上述活化菌体分别接种到里面, 对照组没有接入任何菌体, 所有处理包括对照组都进行3组平行样, 30℃下恒温培养.在加入菌体0、5、13、24、29、35、48、60、72、83、96、120、144、168 h时采集混悬液0.22 μm, 过滤测定液相中As(Ⅲ)和As(Ⅴ), 在未加入菌体0、6、12、24、48、55、72、84、96、120、144、168 h采集混悬液0.22 μm, 过滤测定液相中As(Ⅲ)和As(Ⅴ), 并分别在取样同时检测体系中的Eh及pH.培养结束后, 将溶液离心, 在厌氧箱中倒掉上清液, 收集沉淀物, 用冷冻干燥机(LGJ冷冻干燥机, 北京松源华兴科技发展有限公司)干燥后保存, 一部分用于X射线衍射(XRD, 德国Bruker公司)分析和场发射扫描电镜(FE-SEM, SU-8 000, Hitachi)观察.另一部分用于X射线吸收近边结构[XANES, 台湾同步辐射光源(NSRRC)]分析.
1.6 溶液样品的分析与测定将定期采集的混悬液用0.22 μm的滤膜过滤后, 采用高效液相色谱-原子荧光光谱仪(AFS-8130, 吉天, 中国)测定溶液中的砷形态和含量. HPLC-AFS对As(Ⅲ)和As(Ⅴ)的检出限分别为0.7μg·L-1和1.7 μg·L-1.溶液中Eh的测定使用氧化还原电位测定仪(Thermo Scientific, 美国).溶液中pH的测定使用pH测定仪(Mettler Toledo, 上海).
1.7 实验表征 1.7.1 X射线衍射分析(XRD)固体样品XRD表征使用德国Bruker公司D8 Focus型多晶X射线衍射仪, 采用Cu靶陶瓷X光管.样品置于单晶硅片上铺平, 厚度为0.5 mm, 扫描范围为2θ从15°~90°.数据分析使用MDI Jade 6软件, 利用粉末衍射联合会国际数据中心(JCPDS-ICDD)提供的各种物质标准粉末衍射资料(PDF)进行对照分析.相对结晶度用样品的衍射峰面积与参比样衍射峰面积的百分比值表示[34], 用实验室制备的针铁矿作为参比样.
1.7.2 FE-SEM表征SEM表征使用Hitachi公司SU-8000, 冷场发射枪, 加速电压:0.5~30 kV, 分辨率:1.5 nm(15 kV), 5 nm(1 kV), 能谱可鉴定的元素成分:Be-U, 分辨率:137 eV.
1.7.3 X射线吸收近边结构(XANES)固体样品在真空下冷冻干燥, 密封在两层卡普顿胶带之间进行分析.在台湾同步辐射装置(NSRRC)的01C1波束线上采集了As的K边(11 867 eV)从-150~300 eV的XANES光谱.同时分析了Na3AsO4、Na3AsO3、As2S3和Goethite-As(Ⅴ)标品.在IFEFFIT计算机软件包中用Athena程序对光谱进行分析, 进行能量标定、多次扫描平均、背景减法、归一化和线性拟合.在拟合过程中不允许能量转移, 单个砷形态的权重被限制在0~1之间, 所有组分之和被限制为1.
1.8 数据分析本研究数据分析采用软件SPSS Statistics(version 20.0, IBM)和OriginPro 8进行, 独立样品t检验用于判定处理与对照之间是否存在显著差异.当P < 0.05时, 认为是显著相关.
2 结果与讨论 2.1 砷的释放及还原As的形态及浓度变化如图 1(a)所示, 未加菌的对照组中As(Ⅴ)逐渐释放进水体, 最终在60 h后达到大概6 μmol·L-1后趋于稳定.然而在DP4中, 第13 h砷的释放量达到最大值(12.6 μmol·L-1), 占总吸附量的79%, 是对照组第13 h释放量(1.5 μmol·L-1)的8.4倍. 13 h之后, As(Ⅴ)浓度逐渐降低, 84 h后DP4体系中的As(Ⅴ)的浓度低于空白对照.作者推测初期砷大量释放是由于铁矿物和吸附态砷形态的改变造成吸附态五价砷的释放, 此过程与体系中pH、Eh的变化密切相关[21, 35].后期砷的再次固定可能是由于砷硫矿物的生成或新生的二次铁矿对砷的再吸附[23, 36], 为了验证这个猜测, 其后对固相进行了表征.

|
(a)两个体系中As(Ⅴ)、As(Ⅲ)随时间的变化; (b)砷释放的百分比随时间的变化 图 1 菌株DP4处理下As浓度及形态的变化 Fig. 1 Change of As concentrations and species as a function of the incubation time mediated by DP4 |
另一方面, 对照组和DP4中, 溶解态砷的形态主要是以As(Ⅴ)为主, 没有检测到As(Ⅲ)的存在.出现这种现象有两种可能, 一是DP4还原了溶解态As(Ⅴ)至As(Ⅲ)后, As(Ⅲ)又全部吸附至固相; 二是菌株DP4不能还原溶解态砷.对于第一个可能性, 因为As(Ⅴ)在针铁矿上的吸附力要强于As(Ⅲ), 在溶液中有大量As(Ⅴ)的情况下, As(Ⅲ)不可能全部再吸附到固相中[37].因此, 推测在该实验体系中, DP4没有还原溶解态砷.这一现象与之前的研究结果相悖.在之前的研究工作中, DP4与吸附态As(Ⅴ)共存时, 会释放出As(Ⅲ), 即DP4可将As(Ⅴ)还原为As(Ⅲ), 并释放入水体[18].造成此差异可能的原因是吸附载体的不同, 之前的研究中As是吸附在稳定的二氧化钛上, 而此体系中是吸附在铁氧化物上.此结果说明铁氧化物的存在可能阻碍了砷还原的过程[23].
2.2 pH和Eh的变化pH值随时间的变化如图 2所示, 在非生物对照中, pH值保持在7.60左右, 几乎没有变化.加入菌株DP4的实验中, 溶液中的pH值从最初的7.63增加到7.80.这是由于DP4将乳酸根氧化生成乙酸, 生成CO2, 增加了溶液的碱度造成的[38]. Smedley等[8]发现地下水中的砷浓度与pH值成正相关性的规律, 从酸性到中性条件下基本没有吸附态砷释放; 当pH值增加, 特别是pH>8.50时, 显著促进砷的释放.在该实验体系中, pH值随时间的波动在7.60~7.80之间变化, 对砷释放量的影响不大, 因而在该实验体系中, pH值并非影响砷释放的主要因素.

|
图 2 pH随时间的变化 Fig. 2 Change of the pH as a function of the incubation time |
Eh值随时间的变化如图 3(a)所示, 在对照组中, Eh值维持在110~160 mV之间, 基本处于稳定环境.而DP4中, 在前13 h, Eh从-175.7 mV上升到-157.4 mV左右, 在13 h之后, Eh从-157.4 mV降低到-311 mV左右. Cruz等[38]发现加入SRB后, Eh从原来的(230±50)mV降低到(-310±50)mV, 与本实验结果一致.

|
(a)两个体系中Eh随时间的变化; (b)DP4体系总砷浓度及Eh随时间的变化 图 3 菌株DP4处理下Eh和总砷浓度随时间的变化 Fig. 3 Change of Eh and the concentrations of total dissolved As as a function of the incubation time mediated by DP4 |
有趣的是, 加入菌株DP4体系中, 总砷的浓度同Eh的变化显著相关(P=0.001), 因而推测Eh可能对砷的释放影响较大[图 3(b)]. Carraro等[21]的研究也发现, 在氧化条件下, 砷较易被吸附; 当Eh在-100 mV和-215 mV之间, 吸附态砷易被释放; 当Eh小于-215 mV时, 溶解态砷被再次固定, 生成砷-硫矿物或者与硫铁矿共沉淀[21, 36].此体系中, DP4中的Eh低于-215 mV, 是否生成了砷-硫矿或硫铁矿, 为此对固相进行了研究.
2.3 固相的变化首先通过XRD对吸附载体针铁矿的形态结构变化进行表征, 如图 4所示, 其相对结晶度变化如表 1所示.两体系中有晶形结构的铁相仍以针铁矿为主.不同之处在于, 对照组XRD谱图中针铁矿的峰强度相较初始针铁矿变化不大(109%), 说明培养基本身对针铁矿的结晶度影响不大.而DP4中的针铁矿的峰强度相较初始针铁矿有显著降低(50%), 说明在DP4作用下, 针铁矿向无定形铁矿转化.一般而言, 矿物结晶度越低, 吸附砷能力越强[39].这可能是DP4中在13 h后溶解态砷再次固定至固相的原因[图 1(a)].

|
图 4 固体样品的XRD谱图 Fig. 4 XRD spectra of solid samples |
|
|
表 1 固体样品相对于初始针铁矿的相对结晶度 Table 1 Relative crystallinity of solid samples |
进一步通过扫描电子显微镜SEM(图 5)对固相形貌变化进行表征, 发现在菌株DP4作用下, 针铁矿会发生团聚, 可能是由于微生物分泌物的黏蛋白诱导的[40].此外, 通过SEM-EDS的结果分析中发现, 初始针铁矿[图 5(a)]和对照组[图 5(b)]固相中, 元素组成以铁(Fe)和氧(O)为主, 元素硫(S)的原子含量均低于1%, 与针铁矿分子式[FeO(OH)]一致.然而, 在加入DP4的体系中, S的原子含量显著增多(12%), 可能是生成硫化砷或者硫化铁.如果生成的只是硫化砷, 根据吸附态砷的含量(1.2 g·kg-1)和其方程式As2S3/AsS, 其固相硫的原子含量应为0.08%左右.由此可见, DP4中高达12%的硫含量, 主要是由于硫铁矿的生成.

|
图中红色方框部分是做EDX的部分 图 5 两体系的固体样品及原始样品的SEM图及EDX分析结果 Fig. 5 SEM images and EDX of solid samples of the two systems and the original sample |
为了进一步验证固相中是否生成了硫化砷沉淀, As K-edge XANES光谱用于分析固体上砷的形态, 结果如图 6和表 2所示.对照组中, 固体中As(Ⅴ)含量占100%, 与初始针铁矿吸附状态一致.在DP4的体系中, 没有检测到硫化砷的存在.之前的研究结果表明, 只有As(Ⅲ)/S(摩尔比, 下同)≥0.22时才会生成砷-硫矿物, 当As(Ⅲ)/S < 0.03时则不会生成硫化砷矿物[18].而在本实验研究中As(Ⅲ)/S低于0.03, 所以没有砷化硫生成.另一方面, 有研究也表明[23], S2-优先与铁相反应生成硫化铁(即FeS或FeS2)作为二次沉淀物, 之后, As(Ⅲ)则可与新生成的二次沉淀物通过吸附或共沉淀被再次固定.因此, 在本实验体系中, 硫化砷的生成不仅受As(Ⅲ)/S摩尔比的限制, 铁相的存在也是阻碍其生成的重要原因之一.

|
圆点是原始数据, 实线是线性拟合 图 6 固相上砷K边的XANES分析 Fig. 6 Observed and linear combination-fitted XANES spectra for the K edge of As for solid samples |
|
|
表 2 固相上砷的XANES光谱的线性组合拟合结果1) Table 2 Linear fit results for the XANES spectra for arsenic in the solid phase |
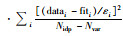 , 式中, Nidp表示模型拟合中的独立点数, Npts表示数据点总数, Nvar表示拟合中的变量数, εi表示不确定性
, 式中, Nidp表示模型拟合中的独立点数, Npts表示数据点总数, Nvar表示拟合中的变量数, εi表示不确定性此外, 虽然没有检测到溶解态As(Ⅲ)(图 1), 固相中却存在19%的As(Ⅲ).因而推测DP4可以原位固相还原As(Ⅴ).之前关于微生物与吸附态砷还原的相互作用研究中, 液相固相均检测到还原的As(Ⅲ), 因而较难判断微生物是否原位还原吸附态砷[18, 19, 41].而关于单一SRB作用下是原位固相还原还是溶解态还原鲜有提及.本实验第一次提供了直接证据证明了硫还原菌可以原位还原吸附态As(Ⅴ)至As(Ⅲ)的过程.
3 结论(1) 本文研究了硫酸盐还原菌DP4介导下, 对吸附在针铁矿上的砷的迁移转化的影响.结果发现, 前期DP4促进了砷的释放, 而后期抑制了砷的释放, 砷的释放与氧化还原电位显著相关.
(2) 砷的释放量减少不是由于与S耦合生成硫化砷物质沉淀, 而是再次吸附到二次铁矿上.
(3) 吸附载体针铁矿形态的改变是影响砷再次吸附的关键因素.在DP4作用下, 针铁矿的结晶度降低了50%, 且发生明显团聚.
(4) 由于溶解态中没有检测到As(Ⅲ)的存在, 而固相中却检测到19%的As(Ⅲ), 推测DP4能原位固相还原As(Ⅴ).
| [1] | Mitchell V L. Health risks associated with chronic exposures to arsenic in the environment[J]. Reviews in Mineralogy and Geochemistry, 2014, 79(1): 435-449. DOI:10.2138/rmg.2014.79.8 |
| [2] | Jing C Y, Liu S Q, Meng X G. Arsenic remobilization in water treatment adsorbents under reducing conditions:Part Ⅰ. Incubation study[J]. Science of the Total Environment, 2008, 389(1): 188-194. DOI:10.1016/j.scitotenv.2007.08.030 |
| [3] | Maillot F, Morin G, Juillot F, et al. Structure and reactivity of As(Ⅲ)-and As(Ⅴ)-rich schwertmannites and amorphous ferric arsenate sulfate from the Carnoulès acid mine drainage, France:comparison with biotic and abiotic model compounds and implications for As remediation[J]. Geochimica et Cosmochimica Acta, 2013, 104: 310-329. DOI:10.1016/j.gca.2012.11.016 |
| [4] | Mohan D, Pittman C U Jr. Arsenic removal from water/wastewater using adsorbents-A critical review[J]. Journal of Hazardous Materials, 2007, 142(1-2): 1-53. DOI:10.1016/j.jhazmat.2007.01.006 |
| [5] | Smedley P L, Kinniburgh D G. A review of the source, behaviour and distribution of arsenic in natural waters[J]. Applied Geochemistry, 2002, 17(5): 517-568. DOI:10.1016/S0883-2927(02)00018-5 |
| [6] | Pichler T, Veizer J, Hall G E M. Natural input of arsenic into a coral-reef ecosystem by hydrothermal fluids and its removal by Fe(Ⅲ) oxyhydroxides[J]. Environmental Science & Technology, 1999, 33(9): 1373-1378. |
| [7] | Yang C L, Li S Y, Liu R B, et al. Effect of reductive dissolution of iron (hydr)oxides on arsenic behavior in a water-sediment system:first release, then adsorption[J]. Ecological Engineering, 2015, 83: 176-183. DOI:10.1016/j.ecoleng.2015.06.018 |
| [8] | Ma J, Guo H, Lei M. Disparity of adsorbed arsenic species and fractions on the soil and soil colloids[J]. Procedia Earth & Planetary Science, 2017, 17: 642-645. |
| [9] | Muehe E M, Scheer L, Daus B, et al. Fate of arsenic during microbial reduction of biogenic versus Abiogenic As-Fe(Ⅲ)-mineral coprecipitates[J]. Environmental Science & Technology, 2013, 47(15): 8297-8307. |
| [10] | Langumier M, Sabot R, Obame-Ndong R, et al. Formation of Fe(Ⅲ)-containing mackinawite from hydroxysulphate green rust by sulphate reducing bacteria[J]. Corrosion Science, 2009, 51(11): 2694-2702. DOI:10.1016/j.corsci.2009.07.001 |
| [11] | Ye L, Liu W J, Shi Q T, et al. Arsenic mobilization in spent nZVI waste residue:effect of Pantoea sp. IMH[J]. Environmental Pollution, 2017, 230: 1081-1089. DOI:10.1016/j.envpol.2017.07.074 |
| [12] | Sun J, Quicksall A N, Chillrud S N, et al. Arsenic mobilization from sediments in microcosms under sulfate reduction[J]. Chemosphere, 2016, 153: 254-261. DOI:10.1016/j.chemosphere.2016.02.117 |
| [13] | Guo H M, Liu Z Y, Ding S S, et al. Arsenate reduction and mobilization in the presence of indigenous aerobic bacteria obtained from high arsenic aquifers of the Hetao basin, Inner Mongolia[J]. Environmental Pollution, 2015, 203: 50-59. DOI:10.1016/j.envpol.2015.03.034 |
| [14] | Polizzotto M L, Harvey C F, Sutton S R, et al. Processes conducive to the release and transport of arsenic into aquifers of Bangladesh[J]. Proceedings of the National Academy of Sciences of the United States of America, 2005, 102(52): 18819-18823. DOI:10.1073/pnas.0509539103 |
| [15] | Rittle K A, Drever J I, Colberg P J S. Precipitation of arsenic during bacterial sulfate reduction[J]. Geomicrobiology Journal, 1995, 13(1): 1-11. DOI:10.1080/01490459509378000 |
| [16] | Ohtsuka T, Yamaguchi N, Makino T, et al. Arsenic dissolution from Japanese paddy soil by a dissimilatory arsenate-reducing bacterium Geobacter sp. OR-1[J]. Environmental Science & Technology, 2013, 47(12): 6263-6271. |
| [17] | Huang J H, Voegelin A, Pombo S A, et al. Influence of arsenate adsorption to ferrihydrite, goethite, and boehmite on the kinetics of arsenate reduction by Shewanella putrefaciens strain CN-32[J]. Environmental Science & Technology, 2011, 45(18): 7701-7709. |
| [18] | Luo T, Tian H X, Guo Z, et al. Fate of Arsenate adsorbed on Nano-TiO2 in the presence of sulfate reducing bacteria[J]. Environmental Science & Technology, 2013, 47(19): 10939-10946. |
| [19] | ThomasArrigo L K, Mikutta C, Lohmayer R, et al. Sulfidization of organic freshwater flocs from a minerotrophic peatland:speciation changes of iron, sulfur, and arsenic[J]. Environmental Science & Technology, 2016, 50(7): 3607-3616. |
| [20] | Rodriguez-Freire L, Moore S E, Sierra-Alvarez R, et al. Arsenic remediation by formation of arsenic sulfide minerals in a continuous anaerobic bioreactor[J]. Biotechnology and Bioengineering, 2016, 113(3): 522-530. DOI:10.1002/bit.v113.3 |
| [21] | Carraro A, Fabbri P, Giaretta A, et al. Effects of redox conditions on the control of arsenic mobility in shallow alluvial aquifers on the Venetian Plain (Italy)[J]. Science of the Total Environment, 2015, 532: 581-594. DOI:10.1016/j.scitotenv.2015.06.003 |
| [22] | Rodriguez-Freire L, Sierra-Alvarez R, Root R, et al. Biomineralization of arsenate to arsenic sulfides is greatly enhanced at mildly acidic conditions[J]. Water Research, 2014, 66: 242-253. DOI:10.1016/j.watres.2014.08.016 |
| [23] | Fan L J, Zhao F H, Liu J, et al. The As behavior of natural arsenical-containing colloidal ferric oxyhydroxide reacted with sulfate reducing bacteria[J]. Chemical Engineering Journal, 2018, 332: 183-191. DOI:10.1016/j.cej.2017.09.078 |
| [24] | Smedley P L, Nicolli H B, Macdonald D M J, et al. Hydrogeochemistry of arsenic and other inorganic constituents in groundwaters from La Pampa, Argentina[J]. Applied Geochemistry, 2002, 17(3): 259-284. |
| [25] | Stuc kman M Y, Corrigan L N, Lenhart J J. Biotic arsenic release from spent adsorbents under anaerobic landfill conditions[J]. Applied Geochemistry, 2017, 78: 321-333. DOI:10.1016/j.apgeochem.2017.01.002 |
| [26] | Moon H S, Kim B A, Hyun S P, et al. Effect of the redox dynamics on microbial-mediated As transformation coupled with Fe and S in flow-through sediment columns[J]. Journal of Hazardous Materials, 2017, 329: 280-289. DOI:10.1016/j.jhazmat.2017.01.034 |
| [27] | Le Pape P, Battaglia-Brunet F, Parmentier M, et al. Complete removal of arsenic and zinc from a heavily contaminated acid mine drainage via an indigenous SRB consortium[J]. Journal of Hazardous Materials, 2017, 321: 764-772. DOI:10.1016/j.jhazmat.2016.09.060 |
| [28] | Xu L Y, Wu X, Wang S F, et al. Speciation change and redistribution of arsenic in soil under anaerobic microbial activities[J]. Journal of Hazardous Materials, 2016, 301: 538-546. DOI:10.1016/j.jhazmat.2015.09.030 |
| [29] | Kim M J, Nriagu J, Haack S. Arsenic species and chemistry in groundwater of southeast Michigan[J]. Environmental Pollution, 2002, 120(2): 379-390. |
| [30] | Savage K S, Tingle T N, O'Day P A, et al. Arsenic speciation in pyrite and secondary weathering phases, Mother Lode Gold District, Tuolumne County, California[J]. Applied Geochemistry, 2000, 15(8): 1219-1244. DOI:10.1016/S0883-2927(99)00115-8 |
| [31] | Couture R M, Rose J, Kumar N, et al. Sorption of arsenite, arsenate, and thioarsenates to iron oxides and iron sulfides:a kinetic and spectroscopic investigation[J]. Environmental Science & Technology, 2013, 47(11): 5652-5659. |
| [32] | Schwertmann U, Cornell R M. Iron oxides in the laboratory:preparation and characterization[M]. 2nd ed.. Weinheim, Germany: Wiley-VCH, 2000. |
| [33] | 刘杰, 张程程. 破解土壤砷污染困局[J]. 瞭望, 2008(32): 17-18. |
| [34] | 尹建军, 邢伟静, 李玉波, 等. ZSM-5分子筛结晶度及晶粒大小的影响因素[J]. 分子催化, 2012, 26(2): 162-168. |
| [35] | Johnston S G, Burton E D, Keene A F, et al. Arsenic mobilization and iron transformations during sulfidization of As(Ⅴ)-bearing jarosite[J]. Chemical Geology, 2012, 334: 9-24. DOI:10.1016/j.chemgeo.2012.09.045 |
| [36] | Serrano J, Leiva E. Removal of arsenic using acid/metal-tolerant sulfate reducing bacteria:a new Approach for bioremediation of high-arsenic acid mine waters[J]. Water, 2017, 9(12): 994. DOI:10.3390/w9120994 |
| [37] | Amstaetter K, Borch T, Larese-Casanova P, et al. Redox transformation of arsenic by Fe(Ⅱ)-activated goethite(α-FeOOH)[J]. Environmental Science & Technology, 2010, 44(1): 102-108. |
| [38] | Cruz Viggi C, Pagnanelli F, Cibati A, et al. Biotreatment and bioassessment of heavy metal removal by sulphate reducing bacteria in fixed bed reactors[J]. Water Research, 2010, 44(1): 151-158. DOI:10.1016/j.watres.2009.09.013 |
| [39] | Jambor J L, Dutrizac J E. Occurrence and constitution of natural and synthetic ferrihydrite, a widespread iron oxyhydroxide[J]. Chemical Reviews, 1998, 98(7): 2549-2585. DOI:10.1021/cr970105t |
| [40] | Mathoera R B, Kok D J, Visser W J, et al. Cellular membrane associated mucins in artificial urine as mediators of crystal adhesion:an in vitro enterocystoplasty model[J]. The Journal of Urology, 2001, 166(6): 2329-2336. DOI:10.1016/S0022-5347(05)65581-4 |
| [41] | Burton E D, Johnston S G, Kocar B D. Arsenic mobility during flooding of contaminated soil:the effect of microbial sulfate reduction[J]. Environmental Science & Technology, 2014, 48(23): 13660-13667. |