 2018, Vol. 39
2018, Vol. 39



N2O是一种重要的温室气体, 其增温潜势是CO2的100~310倍[1], 对全球温室效应的贡献约占5%~6%, 废水生物处理过程是N2O的主要产生源之一[2, 3].导致N2O产生的因素有:溶解氧浓度、亚硝酸盐浓度、C/N比, 温度和有毒物质等[4, 5].短程硝化与传统脱氮工艺相比, 可以减少25%的曝气量, 40%的外加碳源[6].因此, 其成功应用于市政污水和工业废水等的处理[7, 8].虽然短程硝化工艺有工程应用的潜力, 但是亚硝积累是促进N2O排放的主要因素[9], 因为高的亚硝酸盐浓度抑制了N2O还原酶的活性[10, 11].因此, 为了控制和减少短程硝化过程N2O的释放量, 影响N2O释放的关键因素需严格调控.在污水生物脱氮过程中, 主要通过3种途径产生N2O, 分别为:①羟胺(NH2OH)氧化过程, 在硝化过程中, 硝化细菌将NH2OH氧化为NO2-产生副产物N2O, 该过程是在NH2OH氧化还原酶(HAO)的作用下, NH2OH氧化为N2O.或者亚硝酸盐或氨氧化中间产物羟胺的化学分解作用产生[12, 13]; ②硝化细菌(AOB)反硝化过程, 当硝化过程溶解氧不足或NO2-浓度过高时, NH4+-N氧化为NO2-之后自养硝化细菌会将NO2-还原为N2O, 该过程称为硝化细菌反硝化[5, 14]; ③异养反硝化过程(HET), 当反硝化过程缺少N2O还原酶或脱氮酶不平衡时会导致异养反硝化菌生成N2O.此外, 亚硝酸盐积累或者可生物利用的有机碳源不足导致N2O产生[15].
目前, 研究较多的是不同运行条件对短程硝化过程中N2O释放量的影响, 虽然N2O可能通过以上途径产生, 但是对于不同条件下短程硝化过程N2O的产生途径及各个途径所占的比例研究较少, 本研究采用稳定同位素的方法对N2O产生途径进行分析, 通过批次试验的方式, 分析不同溶解氧条件下短程硝化过程N2O释放量及产生途径, 从其产生来源提出N2O减量化的控制措施.
1 材料与方法 1.1 试验装置本试验装置如图 1所示, 反应器内装有从稳定运行短程SBR反应器内取出的活性污泥, 外加试验所需的模拟废水, 顶部用橡胶塞密封, 并开有5个直径不同的小孔, 分别为DO和pH探头孔及曝气、取水样、集气孔.反应器总体积1 L, 有效容积0.7 L.反应器置于恒温磁力搅拌器上, 维持池体内温度为25℃, 并用磁力转子搅拌, 使池体内处于混匀状态.

|
图 1 短程硝化过程批次试验装置示意 Fig. 1 Batch test device of the partial nitrification process |
批次试验所用污泥取自稳定运行短程SBR内活性污泥, 本批次试验用水为人工配水, 即向蒸馏水中添加所需物质, 根据试验目的不同, 配置相应的微量元素溶液和营养液.配水主要成分(mg ·L-1)为: KH2PO4 27, CaCl2 136, MgSO4 ·7H2O 20, NaHCO3 150, (NH4)2SO4(按需配制).微量元素Ⅰ、Ⅱ浓度为:1.0 mL ·L-1.微量元素Ⅰ组成(g ·L-1)为:EDTA 5, FeSO4 5;微量元素Ⅱ组成(g ·L-1)为:EDTA 15, CuSO4 ·5H2O 0.25, ZnSO4 ·7H2O 0.43, H3BO4 0.014, MnCl2 ·4H2O 0.99, Na2MoO4 ·2H2O 0.22, CoCl2 0.24.
1.3 批次试验运行方法批次试验所用污泥浓度为1.82~1.93 g ·L-1控制温度为(25±1)℃, 进水NH4+-N浓度为25mg ·L-1, 将反应器先后控制在DO浓度为(2.5±0.5)、(1.5±0.2)和(0.5±0.1)mg ·L-1条件下运行, 首先加入所需基质, 然后加入碳酸氢钠调节pH为7.5, 盖上胶塞, 插入DO、pH探头, 打开加热, 从曝气口曝气, 然后将取样口用止水夹卡住, 集气口接集气袋.采用在线监测实时控制反应过程中DO浓度, 每个DO浓度做两组平行.反应0、15、30、60 min, 之后每隔60 min取样到曝气结束, 分别取10 mL水样离心, 过滤后测定NH4+-N、NO2--N、NO3--N及COD.分别取6 mL水样用0.45 μm滤膜过滤, 其后4.5 mL水样用于测定溶解态N2O.
1.4 分析指标与测定方法 1.4.1 水质测定方法COD采用快速消解分光光度法测定; NO2--N采用N-(1-萘基)-乙二胺光度法测定[16], NO3--N采用麝香草酚分光光度法测定[16], NH4+-N采用纳氏试剂分光光度法测定[16]; MLSS采用滤纸重量法测定; Multi340i型便携式多功能pH值和DO测定仪在线测定反应过程中的pH值和DO浓度.COD采用快速消解分光光度法测定; NO2--N采用N-(1-萘基)-乙二胺光度法测定[16], NO3--N采用麝香草酚分光光度法测定[16], NH4+-N采用纳氏试剂分光光度法测定[16]; MLSS采用滤纸重量法测定; Multi340i型便携式多功能pH值和DO测定仪在线测定反应过程中的pH值和DO浓度.
1.4.2 N2O释放量及产生途径分析方法(1) 气态N2O测定
反应装置密闭, 所产气体经干燥管干燥后收集于气体采样袋中.采用Agilent 7890气相色谱仪测定N2O, 所用色谱柱为HP-Plot/分子筛(30 m×0.53 mm内径×25 μm膜), 色谱条件为:进样口110℃, 炉温180℃; ECD检测器300℃, 所有气体样品均经多次测定, 直至重现性较好为止.
(2) 溶解态N2O测定
溶解态N2O采用上部空间法测定[17].在密闭条件下, 取测定水样.为抑制残余微量微生物的活性, 向12 mL顶空瓶中加入0.5 mL浓度为1 000 mg ·L-1的HgCl2溶液, 然后加入4.5 mL经0.45 μm滤膜过滤后的水样.将制好的样品放入恒温磁力搅拌器中振荡1 h, 测定上部气体中的N2O浓度, 根据测得的N2O浓度及亨利定律计算出溶解态N2O浓度.
1.4.3 同位素测定方法采用稳定同位素的方法对N2O产生途径进行分析.与其它技术相比, 稳定同位素技术的优点在于使得生态和环境科学问题的研究能够定量化并且是在没有干扰(如没有放射性同位素的环境危害)的情况下进行.本试验采用Isoprime100稳定同位素质谱分析系统对所集气体进行同位素分析, 分析过程示意如图 2所示.具体步骤如下:抽空20 mL顶空瓶, 立即注入待测气体, 通过自动进样器用约25 mL ·min-1的He气流将待测样品吹进含烧碱石棉的化学阱, 99.99%的CO2被吸收. N2O和其他空气组分被捕集在-196℃的冷阱T2中.吹扫300 s后, T2自动移出液氮罐, 并通过六通阀的转换, 将被分析组分转移至-196℃的T3冷阱内, 转换阀的另一头与色谱柱相连接.待T3移出液氮容器即开始进行GC分析, 之后进行质谱分析, 用99.99%的N2O气体做参考气, 测定结果用反硝化法得到的N2O标准气体校正, 即用USGS32、USGS34和IAEA-N1产生的标准气体校准样品N2O氮的bulk值和α值, 用USGS34, USGS35和IAEA-N1产生的标准气体校准气体N2O氧的同位素值.最终根据所得数据计算出15Nbulk、15Nα及SP值.

|
图 2 Isoprime100稳定同位素质谱N2O分析示意 Fig. 2 Diagram of N2O Isoprime100 stable isotope mass spectrometry analysis |
N2O释放量和溶解态N2O浓度根据Noda提供的方法计算[18], 见式(1).
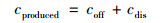
|
(1) |
式中, cproduced为N2O产生量(mg ·L-1); coff为试验过程释放到大气中的N2O浓度(mg ·L-1); cdis为溶解于水中的N2O浓度(mg ·L-1).
N2O的产生途径计算公式[19]见式(2)和(3):
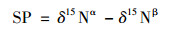
|
(2) |
式中, δ15Nα为15N在中心位(Nα)的丰度(‰), 即14N15N16O; δ15 Nβ为15N在边位(Nβ)的丰度(‰), 即15N14N16O.
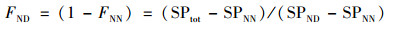
|
(3) |
式中, FND为NH2OH氧化过程产生N2O所占比例(%); FNN为AOB反硝化过程产生N2O所占比例(%); SPNN为NH2OH氧化过程的SP值(‰); SPND为AOB反硝化过程的SP值(‰).
2 结果与讨论 2.1 不同溶解氧条件下短程硝化过程N2O释放量生物脱氮过程中所产生的N2O在系统内的转移与传递主要涉及两个过程:N2O的溶解和N2O的释放. N2O的溶解过程指微生物生成的气态N2O溶解于混合液中, 为气相到液相的传递; N2O的释放过程是指由于曝气吹脱或过饱和等原因溶解态N2O从混合液中释放到大气中, 为液相到气相间的传递.
在不同溶解氧条件下, 对短程硝化过程中溶解态N2O和气态N2O进行监测, 当溶解氧浓度为0.5 mg ·L-1, 测定结果如图 3所示.反应经过240 min, NH4+-N浓度由23.5 mg ·L-1降至2.78 mg ·L-1, 63.5%的NH4+-N转化为NO2--N, 亚硝积累率(NAR)为90.43%, 比氨氧化速率(SAOR, 以N/SS计, 下同)为1.86mg ·(g ·h)-1.从图 3中可以看出, 随着反应进行, 前30 min, 溶解态N2O和气态N2O基本没有检测到, 仅有0.875 mg ·L-1的NO3--N产生, 此过程产生这种现象可能是由于曝气的开始阶段, 微生物进行异养呼吸, 使COD和少量NH4+-N消耗合成自身物质.当反应到30 min之后, 溶解态N2O的浓度逐渐增加, 到120 min达到最大值, 随后趋于平缓并开始下降.前60 min, 气态N2O的浓度增加较少, 到120 min, NO2--N浓度大于5 mg ·L-1, 气态N2O的体积分数快速升高, 反应进行到240 min, 达到320.5×10-6, 利用SPSS软件对NO2--N浓度和N2O浓度与进行Pearson相关系数分析, 得出在0.01水平(双侧)上显著相关, 相关性系数R2=0.947, 显著性P=0.004(P < 0.05), 表明NO2--N浓度对N2O的释放有很大关系.有研究表明[20], 短程硝化过程N2O释放量的急剧上升与体系中NO2--N的积累存在显著的相关性, NO2--N具有生物毒性, NO2--N的积累导致氧化亚氮还原酶的活性降低, 从而引起N2O的释放.

|
图 3 单周期内NH4+-N、NO2--N、NO3--N及N2O变化情况 Fig. 3 Changes in NH4+-N, NO2--N, NO3--N, and N2O over a single cycle |
当溶解氧浓度为1.5 mg ·L-1, 测定结果如图 4所示.反应经过120 min, NH4+-N浓度由22.8 mg ·L-1降至3.46 mg ·L-1, 76.4%的NH4+-N转化为NO2--N, 亚硝积累率为93.43%, 比氨氧化速率为3.35 mg ·(g ·h)-1.从中可以看出, 随着反应进行, 前30 min, 仅有极少的溶解态N2O和气态N2O产生, 当反应到30 min之后, 溶解态N2O浓度逐渐增加, 到120 min达到最大值为0.24 mg ·L-1.前30 min, 气态N2O浓度增加较少, 随后当NO2--N浓度大于3 mg ·L-1, 气态N2O的体积分数快速升高, 到240 min, 达到272.6×10-6. Zeng等[21]的研究指出, 当NO2--N质量浓度大于1~2 mg ·L-1即可导致N2O产生.利用SPSS软件对NO2--N浓度和N2O浓度进行Pearson相关系数分析, 得出在0.01水平(双侧)上显著相关, 相关性系数R2=0.986, 显著性P=0.002(P < 0.05), 表明与溶解氧为0.5 mg ·L-1时一样, 短程硝化过程N2O释放量的急剧上升与体系中NO2--N的积累存在显著的相关性, NO2--N的积累可能是导致N2O释放显著上升的主要原因.

|
图 4 单周期内NH4+-N、NO2--N、NO3--N及N2O变化情况 Fig. 4 Changes in NH4+-N, NO2--N, NO3--N, and N2O over a single cycle |
当溶解氧浓度为2.5 mg ·L-1, 测定结果如图 5所示.反应经过120 min, NH4+-N浓度由24.4 mg ·L-1降至3.38 mg ·L-1, 80.3%的NH4+-N转化为NO2--N, 亚硝积累率为95.43%, 比氨氧化速率为3.75 mg·(g· h)-1, SAOR约为低溶解氧(0.5 mg ·L-1)条件下的2.02倍, 可见较高的溶解氧具有更高的SAOR, 短程硝化反应时间随着溶解氧的增加明显缩短.从图 5中可以看出, 随着反应进行, 前30 min, 基本没有溶解态N2O和气态N2O的产生, 当反应到30 min后, 溶解态N2O浓度逐渐增加, 到120 min达到最大值为0.21 mg ·L-1.前30 min, 气态N2O浓度较少, 当NO2--N浓度大于3 mg ·L-1, 气态N2O的体积分数快速升高, 到240 min, 达到261.3×10-6.可见, 不同DO浓度条件下, N2O的产生量不同, 但总体变化趋势相同.利用SPSS软件对NO2--N浓度和N2O浓度与进行Pearson相关系数分析, 得出在0.01水平(双侧)上显著相关, 相关性系数R2=0.974, 显著性P=0.003(P < 0.05), 表明与低溶解氧条件一致, NO2--N浓度与N2O的释放显著相关.

|
图 5 单周期内NH4+-N、NO2--N、NO3--N及N2O变化情况 Fig. 5 Changes in NH4+-N, NO2--N, NO3--N, and N2O over a single cycle |
可见, 不同的溶解氧条件下, 均有一定浓度的N2O产生, NO2--N浓度又是N2O产生的主要影响因素.而短程硝化过程不可避免地导致亚硝酸盐积累, 短程硝化工艺有着广泛的应用前景, 现在已经在一些污水处理厂得到广泛应用并取得了良好的效果.通过短程硝化结合厌氧氨氧化工艺实现一体化, 可以控制亚硝酸盐浓度在较低的水平, 此外, 对于两段式短程硝化-厌氧氨氧化工艺, 可以通过控制半短程硝化, 即一半的氨氮氧化为亚硝酸盐, 进入厌氧氨氧化反应器内, 可以减少亚硝酸盐浓度, 从而减少N2O的产生.因此, 通过设计合理的工艺来控制反应器中的亚硝酸盐浓度, 对控制N2O的总产量有着重要的作用.
不同溶解氧条件, 短程硝化过程N2O释放量如图 6所示.溶解氧浓度分别为0.5、1.5、2.5 mg ·L-1时, 处理1 g NH4+-N释放的N2O量分别为51.7、34.64和28.10 mg.随着溶解氧的升高, 去除单位质量浓度NH4+-N后N2O的释放量有逐渐降低的趋势, 可见, DO浓度对N2O的产生与释放有重要的影响, DO浓度在0.5 mg ·L-1将导致N2O产生量升高, 而较高的DO浓度有利于降低N2O的产生量.刘秀红等[22]采用SBR反应器, 研究不同的溶解氧浓度对生物脱氮过程中N2O产量的影响, 结果表明, 当溶解氧浓度大于1.5 mg ·L-1后, 随着溶解氧浓度的升高N2O产量开始迅速降低.本研究溶解氧大于1.5 mg ·L-1时, N2O的释放量也开始降低, 与其研究结果相同.巩有奎等[23]对短程反硝化过程中N2O的产生机制进行了分析, 结果表明高浓度的溶解氧对N2O还原酶具有较强的毒性, 会抑制N2O的进一步还原.

|
图 6 去除单位质量浓度NH4+-N后N2O的释放量 Fig. 6 Release of N2O after removal of unit mass concentration NH4+-N |
分析图 7给出的数据可知, 当溶解氧为0.5、1.5、2.5 mg ·L-1时, N2O的释放量占进水总氮的比例分别为4.35%、3.27%、2.63%, 由数据可知, 当溶解氧为0.5 mg ·L-1时, N2O的释放量占进水总氮的比例最大, 而当溶解氧为2.5 mg ·L-1时, N2O的释放量占进水总氮的比例最小.随着溶解氧浓度的升高, N2O的释放量占进水总氮的比例逐渐减少. Law等[24]在小试短程硝化SBR反应器内处理合成废水, 控制溶解氧浓度为2.5~3 mg ·L-1, 得出N2O的释放量占进水总氮的比例为1%. Kampschreur等[4]在中试连续流SBR反应器内处理废水, 控制溶解氧浓度为1.0 mg ·L-1, 得出N2O的释放量占进水总氮的比例为3.4%. Desloover等[25]研究短程硝化过程中N2O的产生机制, 控制溶解氧浓度为0.2~0.4 mg ·L-1, 得出N2O的释放量占进水总氮比例为5.1%~6.6%, 其中45%~47%的NH4+-N氧化为NO2--N, 13%~15%的NH4+-N氧化为NO3--N.可见, 随着溶解氧的升高, N2O的释放量占进水总氮的比例逐渐减少, 此外, 由于工艺类型和运行条件不同, 不同研究者得出N2O的释放量占进水总氮比例也不同.溶解氧浓度不同而产生N2O释放量差异的原因可能是由于高溶解氧条件下, AOB利用亚硝酸盐进行好氧反硝化作用减弱, 而溶解氧浓度较低时通常会促进AOB反硝化反应而积累N2O[25].基于以上试验结果, 从提高污水脱氮效率, 降低N2O产生量来考虑, 短程硝化过程控制溶解氧在2.5 mg ·L-1既可以提高比氨氧化速率, 又可以减少N2O的产生.

|
图 7 N2O的释放量占进水总氮的百分比 Fig. 7 Percentage of N2O released to total influent nitrogen |
现有的大量研究表明, 污水生物脱氮过程中微生物的硝化及反硝化代谢过程是污水处理中N2O的主要产生途径, 如表 1所示.
|
|
表 1 污水生物脱氮过程已知的N2O产生与消耗途径[26] Table 1 Known N2O production and consumption pathways that may be occurring during nitrogen removal in wastewater |
Yoshida等[27]提出可以通过分析N2O分子内15N含量, 推测生物脱氮过程N2O产生途径.通过对稳定同位素比质谱仪将线性结构的N2O气体分子离子化后产生的N2O+和NO+离子片段进行定量分析, 计算15N在α位和β位的丰度, 最终通过SP值判断N2O产生途径.由于N2O的SP值与基质的同位素组成无关, 因此SP值可以作为判断N2O产生途径的有力依据.
当溶解氧为0.5、1.5、2.5 mg ·L-1时, 分别对短程硝化批次试验中收集的气体进行稳定同位素测定, 得到N2O气体的SP值, 从而判断反应过程N2O产生途径, 结果如表 2所示
|
|
表 2 不同溶解氧条件下短程硝化同位素测定结果 Table 2 Results of stable isotopes determination of different DO concentrations |
已有研究表明[19], 假定NH2OH氧化过程生成的N2O气体的SP值(SPNN)平均为28.5‰和硝化细菌反硝化过程生成的N2O气体的SP值(SPND)平均为-2‰.计算结果见表 2, 从中可知, 当溶解氧为0.5 mg ·L-1时, 只有AOB反硝化过程生成N2O.由于低溶解氧可能发生异养菌反硝化作用, 而本研究短程硝化前30 min基本没有检测到N2O, 因此, 没有发生异养菌反硝化作用产生N2O.当溶解氧升至1.5 mg ·L-1时, 有4.52%的N2O通过NH2OH氧化过程生成, AOB反硝化过程生成的N2O占95.48%.继续升高溶解氧到1.5 mg ·L-1时, NH2OH氧化过程生成的N2O比例增加至9.11%, AOB反硝化过程生成的N2O占90.89%. Wunderlin等[19]利用同位素技术研究混合菌系统产生N2O特征得出, 在溶解氧大于1 mg ·L-1和NH4+-N浓度大于10 mg ·L-1时, NH2OH氧化过程产生N2O的比例占N2O产生总量的25%.本研究在溶解氧为1.5 mg ·L-1和2.5 mg ·L-1时, 均有NH2OH氧化过程产生N2O.可见, 溶解氧的改变会影响短程硝化过程N2O的产生途径.随着溶解氧的升高, AOB反硝化过程产生N2O占NH2OH氧化过程产生N2O的比例减少, 但是90%以上的N2O仍然是通过AOB反硝化过程产生.
Okabe等[28]的研究表明如果同时存在NO2--N积累则能诱导AOB利用NO2--N作为电子受体进行反硝化, 而大部分AOB反硝化过程以N2O和NO为反应终产物, 此外, Reino等[29]研究亚硝化颗粒污泥短程硝化得出: NO2--N浓度越高AOB反硝化对N2O释放的贡献越大.本研究在不同溶解氧条件下, 均有不同程度的NO2--N积累, 这是导致AOB反硝化作用产生N2O的重要原因. Yu等[30]的研究表明, 在较高的氨氧化速率条件下, 容易导致NH2OH短暂积累, 通过NOH的化学分解作用产生N2O, 本研究高DO条件下短程硝化污泥具有更高的SAOR, 因此, 随着溶解氧的升高, 会导致NH2OH氧化过程产生N2O的比例增大.综上研究结果, 不同溶解氧条件下, 当NO2--N浓度大于3 mg ·L-1即开始产生N2O, 并且随着NO2--N浓度的增加, AOB反硝化过程产生N2O的量增加, 因此, 避免过高的亚硝酸盐积累, 可以减少N2O的产生.
3 结论(1) 不同溶解氧条件下, N2O的释放均与NO2--N浓度显著相关, 当NO2--N浓度大于2 mg ·L-1, 短程硝化过程开始出现N2O释放, 且N2O的释放量随着NO2--N浓度的增加而增加.
(2) 当溶解氧浓度分别为0.5、1.5、2.5 mg ·L-1, N2O的释放量占进水总氮的比例分别为4.35%、3.27%和2.63%, DO浓度对于N2O的产生与释放有重要的影响, 高DO浓度有利于降低N2O的产生量, DO浓度在0.5 mg ·L-1将导致N2O释放量增加.短程硝化过程控制溶解氧在2.5 mg ·L-1既可以提高比氨氧化速率, 又可以减少N2O的产生.
(3) 溶解氧的改变会影响短程硝化过程N2O的产生途径, 随着溶解氧的升高, AOB反硝化过程产生N2O占NH2OH氧化过程产生N2O的比例减少, 但是90%以上的N2O仍然是通过AOB反硝化过程产生.因此, 避免过高的亚硝酸盐积累, 可以减少N2O的产生.
| [1] | IPCC. Climate Change 2007:The Physical Science Basis (Summary for Policymakers)[M]. Cambridge: Cambridge University Press, 2007: 45-47. |
| [2] | Itokawa H, Hanaki K, Matsuo T. Nitrous oxide production in high-loading biological nitrogen removal process under low COD/N ratio condition[J]. Water Research, 2001, 35(2): 657-664. |
| [3] | Ahn J H, Kwan T, Chandran K. Comparison of partial and full nitrification processes applied for treating high-strength nitrogen wastewaters:microbial ecology through nitrous oxide production[J]. Environmental Science & Technology, 2011, 45(7): 2734-2740. |
| [4] | Kampschreur M J, Van Der Star W R L, Wielders H A, et al. Dynamics of nitric oxide and nitrous oxide emission during full-scale reject water treatment[J]. Water Research, 2008, 42(3): 812-26. DOI:10.1016/j.watres.2007.08.022 |
| [5] | Wunderlin P, Mohn J, Joss A, et al. Mechanisms of N2O production in biological wastewater treatment under nitrifying and denitrifying conditions[J]. Water Research, 2012, 46(4): 1027-1037. DOI:10.1016/j.watres.2011.11.080 |
| [6] | Ge S J, Peng Y Z, Qiu S, et al. Complete nitrogen removal from municipal wastewater via partial nitrification by appropriately alternating anoxic/aerobic conditions in a continuous plug-flow step feed process[J]. Water Research, 2014, 55: 95-105. DOI:10.1016/j.watres.2014.01.058 |
| [7] |
马斌.城市污水连续流短程硝化厌氧氨氧化脱氮工艺与技术[D].哈尔滨: 哈尔滨工业大学, 2012. Ma B. Nitritation and anammox achieved in continous reactors treating sewage[D]. Harbin: Harbin Institute of Technology, 2012. http://cdmd.cnki.com.cn/Article/CDMD-10213-1013035472.htm |
| [8] |
邢丽贞, 郑德瑞, 孔进, 等. 高氨氮废水短程硝化过程中N2O释放试验研究[J]. 环境科学学报, 2016, 36(4): 1260-1265. Xing L Z, Zheng D R, Kong J, et al. N2O emission during short-cut nitrification of high ammonium containing wastewater[J]. Acta Scientiae Circumstantiae, 2016, 36(4): 1260-1265. |
| [9] | Colliver B B, Stephenson T. Production of nitrogen oxide and dinitrogen oxide by autotrophic nitrifiers[J]. Biotechnology Advances, 2000, 18(3): 219-232. DOI:10.1016/S0734-9750(00)00035-5 |
| [10] | Rodriguez-Caballero A, Pijuan M. N2O and NO emissions from a partial nitrification sequencing batch reactor:exploring dynamics, sources and minimization mechanisms[J]. Water Research, 2013, 47(9): 3131-3140. DOI:10.1016/j.watres.2013.03.019 |
| [11] | Jia W L, Liang S, Zhang J, et al. Nitrous oxide emission in low-oxygen simultaneous nitrification and denitrification process:sources and mechanisms[J]. Bioresource Technology, 2013, 136: 444-451. DOI:10.1016/j.biortech.2013.02.117 |
| [12] | Kong Q, Liang S, Zhang J, et al. N2O emission in a partial nitrification system:dynamic emission characteristics and the ammonium-oxidizing bacteria community[J]. Bioresource Technology, 2013, 127: 400-406. DOI:10.1016/j.biortech.2012.10.011 |
| [13] | Zeng R J, Yuan Z G, Keller J, et al. Enrichment of denitrifying glycogen-accumulating organisms in anaerobic/anoxic activated sludge system[J]. Biotechnology and Bioengineering, 2003, 81(4): 397-404. DOI:10.1002/(ISSN)1097-0290 |
| [14] | Du R, Peng Y Z, Cao S B J, et al. Characteristic of nitrous oxide production in partial denitrification process with high nitrite accumulation[J]. Bioresource Technology, 2016, 203: 341-347. DOI:10.1016/j.biortech.2015.12.044 |
| [15] | Harris E, Joss A, Emmenegger L, et al. Isotopic evidence for nitrous oxide production pathways in a partial nitritation-anammox reactor[J]. Water Research, 2015, 83: 258-270. DOI:10.1016/j.watres.2015.06.040 |
| [16] | 国家环境保护总局. 水和废水监测分析方法[M]. 第四版. 北京: 中国环境科学出版社, 2002. |
| [17] | Yang Q, Liu X H, Peng C Y, et al. N2O production during nitrogen removal via nitrite from domestic wastewater:main sources and control method[J]. Environmental Science & Technology, 2009, 43(24): 9400-9406. |
| [18] | Noda N, Kaneko N, Mikami M, et al. Effects of SRT and DO on N2O reductase activity in an anoxic-oxic activated sludge system[J]. Water Science & Technology, 2003, 48(11-12): 363-370. |
| [19] | Wunderlin P, Lehmann M F, Siegrist H, et al. Isotope signatures of N2O in a mixed microbial population system:constraints on N2O producing pathways in wastewater treatment[J]. Environmental Science & Technology, 2013, 47(3): 1339-1348. |
| [20] |
于德爽, 李津, 陆婕. 短程与全程硝化反硝化过程中N2O产量比较[J]. 中国给水排水, 2008, 24(7): 66-69. Yu D S, Li J, Lu J. Comparison of N2O production quantity between complete and shortcut nitrification and denitrification processes[J]. China Water & Wastewater, 2008, 24(7): 66-69. DOI:10.3321/j.issn:1000-4602.2008.07.018 |
| [21] | Zeng W, Wang X D, Li B X, et al. Nitritation and denitrifying phosphorus removal via nitrite pathway from domestic wastewater in a continuous MUCT process[J]. Bioresource Technology, 2013, 143: 187-195. DOI:10.1016/j.biortech.2013.06.002 |
| [22] |
刘秀红, 彭轶, 马涛, 等. DO浓度对生活污水硝化过程中N2O产生量的影响[J]. 环境科学, 2008, 39(3): 660-664. Liu X H, Peng Y, Ma T, et al. Effects of DO concentration on N2O production during nitrification for treating domestic wastewater[J]. Environmental Science, 2008, 39(3): 660-664. DOI:10.3321/j.issn:0250-3301.2008.03.020 |
| [23] |
巩有奎, 王淑莹, 王莎莎, 等. DO对短程反硝化过程中N2O产量的影响[J]. 中南大学学报(自然科学版), 2012, 43(1): 395-400. Gong Y K, Wang S Y, Wang S S, et al. Effect of DO on N2O emission during nitrite denitrification process[J]. Journal of Central South University (Science and Technology), 2012, 43(1): 395-400. |
| [24] | Law Y, Ye L, Pan Y T, et al. Nitrous oxide emissions from wastewater treatment processes[J]. Philosophical Transactions of the Royal Society of the Royalsociety B:Biological Sciences, 2012, 367(1593): 1265-1277. DOI:10.1098/rstb.2011.0317 |
| [25] | Desloover J, Vlaeminck S E, Clauwaert P, et al. Strategies to mitigate N2O emissions from biological nitrogen removal systems[J]. Current Opinion in Biotechnology, 2012, 23(3): 474-482. DOI:10.1016/j.copbio.2011.12.030 |
| [26] | Law Y, Lant P, Yuan Z G. The confounding effect of nitrite on N2O production by an enriched ammonia-oxidizing culture[J]. Environmental Science & Technology, 2013, 47(13): 7186-7194. |
| [27] | Yoshida N, Toyoda S. Constraining the atmospheric N2O budget from intramolecular site preference in N2O isotopomers[J]. Nature, 2000, 405(6784): 330-334. DOI:10.1038/35012558 |
| [28] | Okabe S, Oshiki M, Takahashi Y, et al. N2O emission from a partial nitrification-anammox process and identification of a key biological process of N2O emission from anammox granules[J]. Water Research, 2011, 45(19): 6461-6470. DOI:10.1016/j.watres.2011.09.040 |
| [29] | Reino C, Van Loosdrecht M C M, Carrera J, et al. Effect of temperature on N2O emissions from a highly enriched nitrifying granular sludge performing partial nitritation of a low-strength wastewater[J]. Chemosphere, 2017, 185: 336-343. DOI:10.1016/j.chemosphere.2017.07.017 |