 2018, Vol. 39
2018, Vol. 39


2. 中国科学院广州地球化学研究所, 广州 510640;
3. 华南师范大学环境研究院, 广州 510006
2. Guangzhou Institute of Geochemistry, Chinese Academy of Sciences, Guangzhou 510640, China;
3. Environmental Research Institute, South China Normal University, Guangzhou 510006, China
苯胺是一种重要的有机化工原料和精细化工中间体, 同时也具有强致癌、致畸、致突变效应, 被世界卫生组织国际癌症研究机构列入三类致癌物清单.由于含苯胺废水毒性强、生物降解性差, 通过常规的生化处理系统难以有效处理, 直接排放到水体可能产生环境风险.高级氧化技术为含苯胺等有毒有害有机污染物废水的处理提供了一种有效途径, 其中基于SO4·-的氧化技术是近年来兴起的一种新型高级氧化技术, 对水体中的有机污染物具有较好的氧化去除效果[1, 2].
国内外研究者采用了过渡金属[3]、热[4, 5]、光[6]、碱[7]等多种方式活化PS产生SO4·-氧化降解有机污染物.其中, 过渡金属Fe2+活化PS所需活化能仅为14.8 kcal·mol-1(62 kJ·mol-1), 低于热活化等其他活化方式[8, 9], 因此被认为是最经济有效的活化方法.然而, 游离态的Fe2+在酸性极低或中碱性条件下活化PS效果较差[10, 11]; 同时, Fe2+在活化PS反应初期即被迅速消耗, 导致PS活化持续效果较差[12, 13]; 此外, 反应初始阶段大量游离态的还原性Fe2+对活化产生的SO4·-产生强烈的淬灭作用, 降低了SO4·-氧化降解有机污染物的效率[3].针对上述问题, 近几年有研究者[14~17]尝试采用络合剂络合Fe2+, 通过络合剂对Fe2+的屏蔽效应, 提高Fe2+对pH的适应能力, 并调控Fe2+参与反应的速率, 有效提高了Fe2+持续活化PS氧化降解有机污染物的效率.但是, 该方法仍未能彻底解决Fe2+浓度衰减的问题.因此, 在络合铁离子活化PS体系中, 通过络合作用调控铁离子的同时, 更有效地促进反应体系中累积的Fe3+向Fe2+循环转化, 是进一步提高铁离子持续活化PS氧化能力的关键.为此, 本研究采用具有光敏性的草酸[18]作为Fe3+络合剂, 分析了UV光照通过强化铁离子循环转化来提高PS活化氧化降解苯胺的效果和主要影响因素.
1 材料与方法 1.1 主要试剂草酸高铁铵[(NH4)3Fe(C2O4)3·3H2O]、三氯化铁(FeCl3·6H2O)购自阿拉丁化学试剂有限公司, 过硫酸钠(Na2S2O8)购自天津市科密欧化学试剂有限公司, 1, 10-邻菲罗啉(C12H8N2·H2O)和碘化钾(KI)购自天津市天大化学试剂厂, 所有药品均为分析纯.
1.2 实验仪器高效液相测谱仪(LC-20A, 日本Shimadzu)、紫外可见分光光度计(UV-2310Ⅱ, 上海天美科技有限公司)、低压汞灯(15W, 44μW/cm2, Philips)、pH仪(pH510, 美国Eutech).
1.3 实验方法 1.3.1 Fe2+转化实验反应装置如图 1所示, 将100 mL浓度分别为0.25, 0.5, 0.75 mmol·L-1的草酸高铁铵溶液加入150 mL的石英烧杯中, 将石英烧杯置于低压汞灯下方, 控制液面与光源垂直距离为5 cm, 开启紫外灯反应开始计时, 在取样时间点进行取样并立即测定溶液中的Fe2+浓度.不同pH条件下的Fe2+转化实验与上述过程相似, 并预先使用NaOH (0.1 mol·L-1)和H2SO4 (0.1 mol·L-1)调节溶液pH.

|
图 1 光化学反应实验装置示意 Fig. 1 Photochemical reaction device |
将50 mL所需浓度的草酸高铁铵溶液与25 mL所需浓度的苯胺溶液混合加入到150 mL石英烧杯中, 搅拌混合均匀, 随后向该溶液中加入25 mL所需浓度的PS储备液并使用NaOH (0.1 mol·L-1)和H2SO4 (0.1 mol·L-1)调节混合溶液至所需pH值, 随后置于紫外灯下开启反应.在无草酸高铁铵的实验中, 使用相应体积的蒸馏水代替.
1.4 检测分析方法 1.4.1 检测方法按照Liang等[19]提出的KI显色法测定PS的浓度; 采用邻菲罗啉显色法[20]测定Fe2+的浓度; 采用高效液相色谱法检测苯胺浓度, 测试条件为:反相C-18柱(5 mm×250 mm×4.6 mm), 流动相为乙腈/水(体积比)=55/45, 柱温为40℃, 流速为0.50 mL·min-1, 紫外检测波长为231 nm.
1.4.2 数据分析方法分别采用了准一级反应和二级反应动力学模型[如式(1)、(2)]对苯胺降解过程进行拟合, 分析苯胺降解反应动力学过程, 式中c[苯胺]0和c[苯胺]t分别表示苯胺初始浓度和反应t时间后的浓度, kobs为根据拟合结果得到的表观反应动力学常数, 单位分别为min-1和L·(mol·min)-1.

|
(1) |

|
(2) |
本研究定义PS效率为单位浓度PS所能降低的苯胺浓度, 用ηPS表示, 计算方法如式(3):

|
(3) |
式中, c[苯胺]0和c[苯胺]t分别表示苯胺初始浓度和反应t时间后的浓度, c[PS]0和c[PS]t分别表示PS初始浓度和反应t时间后的浓度.
2 结果与讨论 2.1 UV-Fe(C2O4)33-体系中Fe2+的转化过程图 2显示了4种不同浓度的Fe(C2O4)33-溶液在UV照射条件下Fe2+浓度变化趋势.随着初始Fe(C2O4)33-浓度的增加, 转化生成的Fe2+最高浓度不断升高, 且Fe2+浓度变化过程均呈现快速增加、保持稳定和快速下降这3个阶段, 该结果表明Fe2+在转化生成的同时也不断产生消耗.有研究发现Fe(C2O4)33-发生光化学反应过程会产生H2O2并能与Fe2+发生Fenton反应[如式(4)~(9)][21~23].因此, 推测Fe2+浓度变化过程为:Fe(C2O4)33-持续发生光化学反应不断累积Fe2+, 但C2O42-也不断发生分解, 导致Fe2+转化速率持续降低.而生成的Fe2+因参与Fenton反应或被产生的·OH氧化而不断消耗, 同时Fenton反应产生的·OH也会氧化分解C2O42-, 导致Fe2+的转化速率进一步降低.此外, 由于反应过程中不断产生OH-[如式(9)], 当Fe(C2O4)33-浓度分别为0.5、0.75和1 mmol·L-1时, 反应溶液的pH分别从3逐渐升高到7.06、7.26和7.42, pH的变化也可能影响了Fe2+的转化过程.

|
反应条件:ph=3 图 2 不同初始Fe(C2O4)33-反应体系中Fe2+浓度随时间的变化 Fig. 2 Variations in Fe2+ concentrations in processes with different initial concentrations of Fe(C2O4)33- |
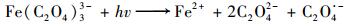
|
(4) |

|
(5) |

|
(6) |

|
(7) |

|
(8) |

|
(9) |
图 3显示了4个不同初始pH的Fe(C2O4)33-溶液中Fe2+浓度变化趋势, Fe(C2O4)33-在酸性条件下具有较好的光化学活性, 在pH为3和5时, Fe2+的转化率分别达到了95%和82%, 而当初始pH为中性及碱性时, Fe2+的转化率显著降低, pH=9时几乎没有Fe2+转化生成.有研究指出C2O42-与Fe3+的络合形态受溶液pH和C2O42-浓度的影响非常显著, 在不同pH条件下, Fe3+在溶液中会以Fe(OH)3、Fe(OH)2+、Fe(C2O4)+、Fe(C2O4)2-、Fe(C2O4)33-等不同形式存在[24].因此, 较高的初始pH可能影响了具有最高光化学活性的Fe(C2O4)33-产生, 从而降低了通过光化学过程生成Fe2+的速率.

|
反应条件:c[Fe(C2O4)33-]0=0.75 mmol·L-1 图 3 不同初始pH条件下Fe(C2O4)33-反应体系中Fe2+转化率 Fig. 3 Variations in Fe2+ conversion efficiency under different initial pH conditions |
图 4显示了5种不同初始Fe(C2O4)33-浓度条件下的PS活化及苯胺氧化降解情况.在无UV光照的Fe(C2O4)33-活化PS体系中, 虽然初始的Fe(C2O4)33-浓度高达5 mmol·L-1, 但PS基本未发生分解.当引入UV光照后, PS的分解率和苯胺的氧化降解效率均显著增强, 且随着初始Fe(C2O4)33-浓度的增加, 由于光化学反应生成的Fe2+浓度逐渐增大, PS的分解率不断提高.当Fe(C2O4)33-初始浓度达到0.75、1、5 mmol·L-1时, PS分别在反应180、150、120 min后可基本被彻底活化分解, 但3个反应体系的苯胺降解率分别为49%、45%、12%, 并未随PS分解速率的增大而升高.此外, 虽然初始Fe(C2O4)33-浓度为0.25 mmol·L-1和0.5 mmol·L-1时的PS分解率分别仅有51%和78%, 但是苯胺的去除率却分别达到了30%和42%, 甚至远高于Fe(C2O4)33-初始浓度为5 mmol·L-1时的苯胺去除率.所有反应体系中苯胺的降解过程(PS未完全分解前)均符合二级反应动力学模型(如表 1), 降解速率常数大小顺序为5 mmol·L-1 < 0.25 mmol·L-1 < mol·L-1 < 1 mmol·L-1 < 0.75 mmol·L-1.而随着Fe(C2O4)33-初始浓度的增加, ηPS依次降低.上述结果一方面是因为在C2O42-络合铁离子活化PS反应体系中, C2O42-与苯胺之间存在对SO4·-的竞争[如式(10)][14], 降低了SO4·-用于氧化降解苯胺的效率, 且这种竞争作用会随着C2O42-浓度的升高而不断增强.同时, 在高浓度Fe(C2O4)33-体系中, 由于生成的Fe2+浓度较高, 虽然活化PS产生更多的SO4·-, 但SO4·-自身以及Fe2+对SO4·-的淬灭作用也会随之加强[如式(11)~(12)][3, 25], 也降低了SO4·-用于氧化降解苯胺的效率.

|
反应条件:pH=3, c[PS]0=5 mmol·L-1, c[苯胺]0=1.25 mmol·L-1 图 4 不同Fe(C2O4)33-浓度反应体系中苯胺和PS浓度的变化曲线 Fig. 4 Variations in aniline and PS concentrations in processes with different Fe(C2O4)33- concentrations |
|
|
表 1 不同Fe(C2O4)33-浓度体系中的苯胺降解速率常数及氧化剂效率(ηPS) Table 1 Rate constants of aniline degradation and oxidant efficiency under various Fe(C2O4)33- concentrations |

|
(10) |

|
(11) |

|
(12) |
考虑到UV可直接光活化分解PS, 因此本研究首先考察了UV光照条件下, 无Fe(C2O4)33-体系中4种不同初始PS浓度条件下UV活化PS氧化降解苯胺的效果, 结果如图 5所示.随着初始PS浓度的增加, 苯胺降解率逐渐增加, 分别为37%、56%、67%、71%; PS活化分解率虽呈现下降, 但活化分解量逐渐增加, PS浓度分别下降了1.04、1.52、1.98、2.44 mmol·L-1.

|
反应条件:pH=3, c[苯胺]0=1.25 mmol·L-1 图 5 不同PS浓度UV-PS体系中苯胺的浓度变化曲线及PS分解率 Fig. 5 Aniline concentration variations and PS decomposition in UV-PS processes with different initial PS concentrations |
苯胺降解过程的反应动力学研究结果显示(如表 2), 在PS初始浓度为5 mmol·L-1和10 mmol·L-1时, 苯胺的降解过程符合二级反应动力学模型, 而当PS浓度进一步增加到15 mmol·L-1和20 mmol·L-1时, 苯胺的降解过程更符合准一级反应动力学模型. Tan等[26]也在研究中发现, 随着PS浓度的降低, 根据一级反应动力学模型拟合出的苯胺降解反应动力学方程的相关系数明显低于高浓度条件下的相关系数.这可能是因为在PS浓度为15 mmol·L-1和20 mmol·L-1时, 由于过高的PS浓度活化产生了足量的SO4·-, 导致影响苯胺氧化降解的因素减少或影响程度降低.另外, 结果显示当PS浓度依次从5 mmol·L-1增加到10、15、20 mmol·L-1时, 苯胺降解率分别增加19%、11%、4%, 苯胺降解率的增加幅度逐渐降低; 对应ηPS分别下降了0.009、0.036、0.062(如表 2), 下降幅度逐渐增加.这可能也是因为当PS浓度大于10 mmol·L-1时, 此时PS已经过量, 并会与反应体系中产生的SO4·-发生反应[如式(13)], 从而降低了SO4·-的利用率.
|
|
表 2 不同反应体系中的苯胺降解反应速率常数及PS效率(ηPS) Table 2 Rate constants of aniline degradation and oxidants efficiency in various processes |

|
(13) |
图 6显示了Fe(C2O4)33-加入后不同PS浓度条件下PS活化及苯胺降解情况.在PS初始浓度分别为5、10、15、20 mmol·L-1条件下, Fe(C2O4)33-加入后PS活化率均显著提高, 分别增加了71%、48%、36%、26%, 苯胺降解率分别增加了12%、6%、5%、4%, 且增加幅度随PS浓度增大逐渐降低.此外, 苯胺降解动力学研究结果显示, 当PS浓度达到10 mmol·L-1时, 苯胺的降解过程与无Fe(C2O4)33-体系不同, 更符合准一级反应动力学模型.上述结果可能是因为当PS浓度达到10 mmol·L-1时, 由于PS活化效果相比无Fe(C2O4)33-的体系得到了进一步强化, 此时已产生了过量的SO4·-. ηPS的计算结果显示, 由于SO4·-的淬灭及竞争反应更加剧烈, UV-Fe(C2O4)33--PS体系中ηPS显著减小, 并且随着初始PS浓度的增加而降低.因此, 综合苯胺降解效果及PS效率等因素, 本研究中较适合的PS浓度为10 mmol·L-1, 且增加PS浓度在氧化降解更高浓度苯胺时应更具有优势.

|
反应条件:pH=3, c[Fe(C2O4)33-]0=0.75 mmol·L-1, c[苯胺]0=1.25 mmol·L-1 图 6 不同PS浓度UV-Fe(C2O4)33--PS体系中苯胺的浓度变化曲线及PS分解率 Fig. 6 Aniline concentration variations and PS decomposition in UV-Fe(C2O4)33--PS processes with different initial PS concentration |
图 7(a)显示了初始pH分别为3、5、7、9时, 反应前180 min的PS浓度变化趋势.在酸性条件下, PS活化效率更高, 这主要归因于酸性条件更有利于UV-Fe(C2O4)33-体系发生光化学反应生成Fe2+.然而, 虽然研究表明pH=3比pH=5更有利于Fe2+生成, 但当反应进行60 min后, 初始pH=5的反应体系中PS活化分解率却略高于pH=3的反应体系.此外, 当初始pH为中碱性条件时, PS仍可被有效活化, 且在反应60 min后, PS的活化速率显著增大.

|
反应条件:c[PS]0=10 mmol·L-1, c[Fe(C2O4)33-]0=0.75 mmol·L-1, c[苯胺]0=1.25 mmol·L-1 图 7 不同初始pH条件下PS浓度和pH变化趋势 Fig. 7 PS concentrations and pH variations in processes with different initial pH values |
对反应体系pH的检测结果显示[如图 7(b)], 由于本研究未对溶液pH进行缓冲控制, 反应过程中溶液pH逐渐下降, 当初始pH分别为3、5、7、9时, pH在反应60 min后分别下降到2.06、2.74、5.07、5.72, 反应120 min时下降到1.87、2.14、2.72、3.82.这是因为PS的活化分解是一个释放H+的反应过程[如式(14)、(15)][27], 且该过程对pH降低的作用远大于UV-Fe(C2O4)33-体系光化学反应过程对pH升高的作用.另外, 有研究指出在较低的pH条件下, Fe(C2O4)33-的稳定性也会受到不利影响[24, 28].由此推断在中碱性条件下, 反应前60 min主要通过UV光活化分解PS, 并导致反应体系pH降低, 从而促进了Fe(C2O4)33-的形成和Fe2+的转化, 最终强化了PS的活化分解.而在初始pH=3时, 由于反应过程中pH进一步降低到强酸性环境, 不利于Fe(C2O4)33-的形成, 而此时pH=5的反应体系更有利于形成Fe(C2O4)33-, 因此在后续反应中PS活化效率更高.

|
(14) |

|
(15) |
为了进一步研究pH的影响, 本研究对反应进行300 min时的剩余PS和苯胺进行了研究, 结果如图 8所示.由于不同初始pH的反应体系在反应30 min后均处于酸性环境, PS活化分解率的差距逐渐缩小, 反应300 min时的剩余PS浓度大小顺序为pH=5 < pH=7 < pH=3 < pH=9, 其中pH=3的反应体系在180~300 min期间, PS活化分解率约为9%, 而pH=5、pH=7、pH=9的反应体系中PS活化分解率分别约为12%、17%、28%, 均超过了pH=3的反应体系.苯胺氧化降解结果显示, pH=7和pH=9反应体系中苯胺降解率分别达到了91%和97%, 超过了pH=3(72%)和pH=5(78%)的反应体系.苯胺的降解过程(pH=7和pH=9的反应体系为反应60 min后)均符合准一级反应动力学模型(如表 3), 降解速率大小顺序为pH=3 < pH=5 < pH=7 < pH=9, 而ηPS大小顺序也符合pH=3 < pH=5 < pH=7 < pH=9.因此, 初始pH为中碱性条件不仅能有效地活化PS氧化降解苯胺, 同时还能有效地提高苯胺对SO4·-的利用效率.这可能是因为在初始pH=3的反应体系中, 由于pH极低, 游离态Fe2+对SO4·-的淬灭反应更加剧烈, 而在较高初始pH反应体系中, SO4·-与OH-反应生成了强氧化性的·OH[如式(16)], 并与SO4·-协同强化了苯胺的氧化降解.

|
反应条件:c[PS]0=10 mmol·L-1, c[Fe(C2O4)33-]0 =0.75 mmol·L-1, c[苯胺]0=1.25 mmol·L-1 图 8 不同初始pH反应体系300 min后的剩余苯胺和PS情况 Fig. 8 Residual aniline and PS (c/c0) after 300 min in processes with different initial pH values |
|
|
表 3 不同pH反应体系中的苯胺降解反应速率常数及氧化剂效率(ηPS) Table 3 Rate constants of aniline degradation and oxidant efficiency in processes with different pH values |

|
(16) |
(1) UV-Fe(C2O4)33-能有效促进Fe2+生成并显著提高PS活化效率, 且酸性条件更有利于生成Fe2+, 并且Fe(C2O4)33-浓度越高, 转化生成的Fe2+最高浓度越大.
(2) Fe(C2O4)33-浓度增加虽然会促进PS活化分解, 但当浓度过高时会降低苯胺降解率.
(3) 在UV-PS体系和UV-Fe(C2O4)33--PS体系中, PS初始浓度增加有利于促进苯胺降解, 但过高的PS浓度对苯胺降解率的提升作用有限, 并且降低了PS的有效利用率.
(4) UV-Fe(C2O4)33--PS体系对初始pH具有较好的适应性, 即使在初始pH为中碱性条件下, PS也可被有效活化, 并且对苯胺表现出更好的去除效果.
| [1] | Devi P, Das U, Dalai A K. In-situ chemical oxidation:principle and applications of peroxide and persulfate treatments in wastewater systems[J]. Science of the Total Environment, 2016, 571: 643-657. DOI:10.1016/j.scitotenv.2016.07.032 |
| [2] |
欧阳磊, 丁耀彬, 朱丽华, 等. 钴掺杂铁酸铋活化过硫酸盐降解水中四溴双酚A的研究[J]. 环境科学, 2013, 34(9): 3507-3512. Ouyang L, Ding Y B, Zhu L H, et al. Efficient degradation of tetrabromobisphenol a in water by Co-doped BiFeO3[J]. Environmental Science, 2013, 34(9): 3507-3512. |
| [3] | Liang C J, Bruell C J, Marley M C, et al. Persulfate oxidation for in situ remediation of TCE. Ⅰ. Activated by ferrous ion with and without a persulfate-thiosulfate redox couple[J]. Chemosphere, 2004, 55(9): 1213-1223. DOI:10.1016/j.chemosphere.2004.01.029 |
| [4] | Huang K C, Couttenye R A, Hoag G E. Kinetics of heat-assisted persulfate oxidation of methyl tert-butyl ether (MTBE)[J]. Chemosphere, 2002, 49(4): 413-420. DOI:10.1016/S0045-6535(02)00330-2 |
| [5] |
蒋梦迪, 张清越, 季跃飞, 等. 热活化过硫酸盐降解三氯生[J]. 环境科学, 2018, 39(4): 1661-1667. Jiang M D, Zhang Q Y, Ji Y F, et al. Degradation of triclosan by heat activated persulfate oxidation[J]. Environmental Science, 2018, 39(4): 1661-1667. |
| [6] | Yang S Y, Wang P, Yang X, et al. Degradation efficiencies of azo dye Acid Orange 7 by the interaction of heat, UV and anions with common oxidants:persulfate, peroxymonosulfate and hydrogen peroxide[J]. Journal of Hazardous Materials, 2010, 179(1-3): 552-558. DOI:10.1016/j.jhazmat.2010.03.039 |
| [7] | Furman O S, Teel A L, Watts R J. Mechanism of base activation of persulfate[J]. Environmental Science & Technology, 2010, 44(16): 6423-6428. |
| [8] | Fordham J W L, Williams H L. The persulfate-iron(Ⅱ) initiator system for free radical polymerizations[J]. Journal of the American Chemical Society, 1951, 73(10): 4855-4859. DOI:10.1021/ja01154a114 |
| [9] | Kolthoff I M, Miller I K. The chemistry of persulfate. I. The kinetics and mechanism of the decomposition of the persulfate ion in aqueous medium1[J]. Journal of the American Chemical Society, 1951, 73(7): 3055-3059. DOI:10.1021/ja01151a024 |
| [10] | 左传梅. Fe(Ⅱ)活化过硫酸盐高级氧化技术处理染料废水研究[D]. 重庆: 重庆大学, 2012. 27-28. http://cdmd.cnki.com.cn/Article/CDMD-10611-1012048228.htm |
| [11] | Xu X R, Li X Z. Degradation of azo dye Orange G in aqueous solutions by persulfate with ferrous ion[J]. Separation and Purification Technology, 2010, 72(1): 105-111. DOI:10.1016/j.seppur.2010.01.012 |
| [12] | Liang C J, Bruell C J, Marley M C, et al. Persulfate oxidation for in situ remediation of TCE. Ⅱ. Activated by chelated ferrous ion[J]. Chemosphere, 2004, 55(9): 1225-1233. DOI:10.1016/j.chemosphere.2004.01.030 |
| [13] | Crimi M L, Taylor J. Experimental evaluation of catalyzed hydrogen peroxide and sodium persulfate for destruction of BTEX contaminants[J]. Soil and Sediment Contamination:An International Journal, 2007, 16(1): 29-45. DOI:10.1080/15320380601077792 |
| [14] | Han D H, Wan J Q, Ma Y W, et al. New insights into the role of organic chelating agents in Fe(Ⅱ) activated persulfate processes[J]. Chemical Engineering Journal, 2015, 269: 425-433. |
| [15] | Dong H Y, Qiang Z M, Hu J, et al. Accelerated degradation of iopamidol in iron activated persulfate systems:roles of complexing agents[J]. Chemical Engineering Journal, 2017, 316: 288-295. DOI:10.1016/j.cej.2017.01.099 |
| [16] | Yu S X, Gu X G, Lu S G, et al. Degradation of phenanthrene in aqueous solution by a persulfate/percarbonate system activated with CA chelated-Fe(Ⅱ)[J]. Chemical Engineering Journal, 2018, 333: 122-131. DOI:10.1016/j.cej.2017.09.158 |
| [17] | Cui H, Gu X G, Lu S G, et al. Degradation of ethylbenzene in aqueous solution by sodium percarbonate activated with EDDS-Fe(Ⅲ) complex[J]. Chemical Engineering Journal, 2017, 309: 80-88. DOI:10.1016/j.cej.2016.10.029 |
| [18] | Kwan C Y, Chu W. The role of organic ligands in ferrous-induced photochemical degradation of 2, 4-dichlorophenoxyacetic acid[J]. Chemosphere, 2007, 67(8): 1601-1611. DOI:10.1016/j.chemosphere.2006.11.052 |
| [19] | Liang C J, Huang C F, Mohanty N, et al. A rapid spectrophotometric determination of persulfate anion in ISCO[J]. Chemosphere, 2008, 73(9): 1540-1543. DOI:10.1016/j.chemosphere.2008.08.043 |
| [20] | Tamura H, Goto K, Yotsuyanagi T, et al. Spectrophotometric determination of iron(Ⅱ) with 1, 10-phenanthroline in the presence of large amounts of iron(Ⅲ)[J]. Talanta, 1974, 21(4): 314-318. DOI:10.1016/0039-9140(74)80012-3 |
| [21] | Jeong J, Yoon J. Dual roles of CO2-· for degrading synthetic organic chemicals in the photo/ferrioxalate system[J]. Water Research, 2004, 38(16): 3531-3540. DOI:10.1016/j.watres.2004.05.016 |
| [22] | Jeong J, Yoon J. pH effect on OH radical production in photo/ferrioxalate system[J]. Water Research, 2005, 39(13): 2893-2900. DOI:10.1016/j.watres.2005.05.014 |
| [23] | Ardila Arias A N, Arriola E, Reyes Calle J, et al. Mineralización de etilenglicol por foto-fenton asistido con ferrioxalato[J]. Revista Internacional de Contaminacion Ambiental, 2016, 32(2): 213-226. |
| [24] | Balmer M E, Sulzberger B. Atrazine degradation in irradiated iron/oxalate systems:effects of pH and oxalate[J]. Environmental Science & Technology, 1999, 33(14): 2418-2424. |
| [25] | Deng J, Shao Y, Gao N, et al. Zero-valent iron/persulfate(Fe0/PS) oxidation acetaminophen in water[J]. International Journal of Environmental Science and Technology, 2014, 11(4): 881-890. DOI:10.1007/s13762-013-0284-2 |
| [26] | Tan C Q, Gao N Y, Zhou S Q, et al. Kinetic study of acetaminophen degradation by UV-based advanced oxidation processes[J]. Chemical Engineering Journal, 2014, 253: 229-236. DOI:10.1016/j.cej.2014.05.013 |
| [27] | Lin C C, Wu M S. Degradation of ciprofloxacin by UV/S2O82- process in a large photoreactor[J]. Journal of Photochemistry and Photobiology A:Chemistry, 2014, 285: 1-6. DOI:10.1016/j.jphotochem.2014.04.002 |
| [28] | Kusic H, Peternel I, Ukic S, et al. Modeling of iron activated persulfate oxidation treating reactive azo dye in water matrix[J]. Chemical Engineering Journal, 2011, 172(1): 109-121. DOI:10.1016/j.cej.2011.05.076 |