 2018, Vol. 39
2018, Vol. 39



2. 中国科学院生态环境研究中心, 环境模拟与污染控制国家重点联合实验室, 北京 100085;
3. 中国科学院城市环境研究所, 中国科学院区域大气环境研究卓越创新中心, 厦门 361021;
4. 中国科学院大学资源与环境学院, 北京 100049
2. State Key Joint Laboratory of Environment Simulation and Pollution Control, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing 100085, China;
3. Center for Excellence in Regional Atmospheric Environment, Institute of Urban Environment, Chinese Academy of Sciences, Xiamen 361021, China;
4. College of Resources and Environment, University of Chinese Academy of Sciences, Beijing 100049, China
二次有机气溶胶(secondary organic aerosol, SOA)是城市地区大气细粒子(PM2.5)的重要组成部分[1, 2], 能够严重影响气候变化、人类健康等[3, 4].它是由人为源和天然源排放的挥发性有机物(volatile organic compounds, VOCs)或半挥发性有机物经过复杂的大气物理化学过程而形成的[5, 6].目前, 已检测到的前体物VOCs就有10 000~100 000种, 他们中绝大部分可以通过大气氧化途径生成SOA[7, 8]. 因此, SOA的化学组成和生成过程成为大气细颗粒物研究最大的难点[9], 其中估算环境大气的SOA生成对于认知SOA生成转化机制及其对大气细粒子污染的影响具有重要意义.
SOA生成潜势(SOA formation potential)是指一定量的VOCs经氧化后能生成的最大SOA的量[10], 可以通过各种方法间接估算, 也可以直接测定. 目前, 国内外间接估算SOA生成潜势的方法主要有:气溶胶生成系数法[11, 12]、气溶胶产率法[13, 14]、挥发性基组法[15, 16]. Kang等[10, 17]发展起来的一种颗粒物氧化流动管反应器(oxidation flow reactor,OFR)可以直接测定SOA的生成潜势, 这种方法将待测VOCs通入含有高浓度羟基自由基(·OH)的OFR内反应, 然后直接利用气溶胶质谱仪(aerosol mass spectrometer, AMS)测定生成的SOA质量浓度, 通过改变·OH浓度使生成的SOA量最大, 此时的SOA生成量即为此VOCs的SOA生成潜势. 相对于烟雾箱系统来说, OFR通过短时间的氧化即可模拟提供几天甚至几周的老化过程, 可以研究VOCs氧化的二代甚至多代反应, 且可以连续观测外场环境大气SOA生成潜势的变化特征. OFR现已被大量应用于实验室研究[18-21], 并且逐步应用于外场观测[22, 23].
城市地区污染情况相对比较复杂.在北京城市地区, 亚微米气溶胶PM1的主要成分是有机物、硫酸盐、硝酸盐、铵盐, 其中有机气溶胶(organic aerosol, OA)占比31%~57%[24]. OA在PM1中的占比随季节变化特征明显, 比如在夏季占~30%, 在冬季占~60%[24]. 但是, SOA在OA中的占比则刚好相反, 夏季为45%~67%, 冬季为22%~50%, 这表明了SOA在夏季OA组成中的重要地位. 夏季与冬季相比有更强烈的光照, 更高浓度的大气氧化剂, 这些因素可能促进了前体物VOCs向SOA的转化, 从而导致夏季SOA在OA中的占比更高.研究夏季SOA的生成潜势, 探索SOA生成潜势的时间变化规律对于理解北京市的SOA污染状况, 控制SOA生成潜势的影响因素, 以及SOA生成对北京细颗粒物污染的影响具有重要意义.本实验利用OFR首次研究了北京市夏季环境大气的SOA生成潜势, 分析了SOA生成潜势的日变化规律, 并且讨论了可能的影响因素.
1 材料与方法 1.1 采样点设置外场实验采样点设置在中国科学院生态环境研究中心综合楼楼顶, 采样点距离双清路200 m, 在奥体中心国控点西北约5 km处, 附近无大的污染点源, 是一个典型的城市采样点.为了减少VOCs的气相损失, 采样管路及连接部分均采用聚四氟乙烯材质, 长度约5 m, 内径2.5 cm. 通过将采样泵采样流量设置为200 L·min-1, 减少采样空气在管路中的停留时间, 最大限度地减少VOCs组分的损失. 在采样泵入口前端引一路采样气进入反应器, 通过反应器出口的尾气泵控制进入反应器的采样气流量为6.6 L·min-1, 其余排空. 采样周期为2017年5月1日至9月30日, 包括整个夏季. 排除维护仪器、校准反应器老化时间等耗费的时间, 用于外场观测的有效时长为112 d, 占整个采样周期的3/4. 采样的时间分辨率为1 h, 总采样时间为2 688 h.
1.2 OFROFR由Kang等[10, 17]首次提出并应用, 通过光解加入反应器的臭氧产生比实际大气高几百倍甚至几千倍的·OH浓度, 其能在非常短的停留时间(几十秒到几分钟)模拟相当于在实际大气环境下几天甚至几周的老化过程, 因此主要用来研究VOCs及SOA的老化过程. 本研究采用由两个体积均为15 L的不锈钢圆筒组成的孪生OFR, 实验系统如图 1所示. OFR内的环境空气与经加湿的臭氧一起进入反应器, 臭氧经臭氧发生器产生, 流量控制在1.2 L·min-1.在反应器出口设置有尾气泵, 其前端通过质量流量计调节经过尾气泵的流量. 本实验中通过反应器的气体总流量为7.8 L·min-1(包括臭氧和环境空气). 经计算, 反应器内流体状态为层流(雷诺数 < 100). 每个OFR长50 cm, 内径20 cm, 且前端安装有一段长30 cm、内径6 cm的混合管以保证采样空气在进入反应器前充分混合, 反应器内壁有一层特氟龙内衬以减少反应器壁的反应性. 两个反应器内均安装有4根254 nm紫外线低压汞灯, 其中一个开启紫外灯作为氧化流化管潜势反应器(称为OFR), 另一个不开紫外灯作为背景对照(称为Bypass反应器). 除此之外, 两个反应器其它条件均保持一致. 反应器内外壁之间通入循环冷却水, 通过调节循环水流量以保证两个反应器内温度都在24~26℃范围内, 并且调节加湿器保证反应器内湿度在30%~40%范围内. 反应器温湿度通过安装在反应器内出口旁的温湿度探头实时测定. 在实验期间, 由电脑控制三通电磁阀切换气体样品间断进入OFR和Bypass反应器, 每30 min切换一次. 由于采样空气在反应器内的停留时间很短(~115 s), 同时通过Bypass反应器的对照, 可以忽略气相和颗粒相的壁损失对SOA生成的影响.

|
图 1 OFR实验系统 Fig. 1 Oxidation flow reactor system |
在254 nm紫外线的照射下, 臭氧和加入的水分会发生如下反应[式(1)和(2)]产生氧化剂·OH:
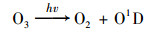
|
(1) |
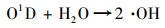
|
(2) |
反应器内老化时间等于·OH浓度乘以反应器平均停留时间, 相当于在实际大气环境中老化的时间, 通过SO2经过OFR前后的衰减量进行估算[22, 25]. 计算方法如式(3)所示.反应器内的·OH浓度计算方法如式(4)所示. 为排除由于紫外灯老化造成的影响, 本研究中, 每月定期用SO2通入反应器测算实验系统的老化时间(aging time, AT).
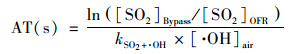
|
(3) |
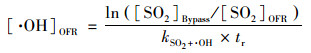
|
(4) |
式中, [SO2]Bypass表示经过Bypass反应器的SO2浓度, [SO2]OFR表示经过OFR的SO2浓度, kSO2+·OH表示SO2与·OH的反应速率常数, [·OH]air表示环境大气中·OH的平均浓度, 本研究中取为1.5×106 molecules·cm-3 [26], tr为环境大气在反应器中的停留时间, 本研究中为115 s.
1.3 仪器高分辨飞行时间气溶胶质谱仪(HR-ToF-AMS, AMS, 美国Aerodyne公司)可以测定非难熔颗粒相的有机物、硫酸盐、硝酸盐、铵盐和氯盐. 本实验中利用AMS测定OFR内生成的SOA, 时间分辨率为3 min. AMS测量的是颗粒物中的非难熔组分. 气溶胶粒子进入AMS到达热蒸发器(温度约873K)后, 沸点在此温度以下的物质能被蒸发后通过EI的方式电离为碎片离子, 再通过质谱检测器检测. 对于有机分子来说, 基本都能被电离并检测到. 将AMS的收集效率设置为0.5[27, 28]. 扫描电迁移率颗粒物粒径谱仪(SMPS, 美国TSI公司, 型号3082)由差分电迁移率分析仪(DMA, 美国TSI公司, 型号3081A)和冷凝颗粒物计数器(SMPS, 美国TSI公司, 型号3776)构成, 用来测定总的PM1质量浓度, 时间分辨率为5 min. 在仪器标定和实验数据的处理中, 利用SMPS和AMS同时测量硝酸铵或者SOA的浓度来校准AMS数值, 减少SOA质量浓度测量的不确定性. 臭氧、一氧化碳、氮氧化物和二氧化硫浓度分别利用美国Thermo Fisher的臭氧分析仪(49i-LTE)、一氧化碳分析仪(48i-TLE)、氮氧化物分析仪(42iTL)和二氧化硫分析仪(43i)进行测定, 时间分辨率为1 min. VOCs浓度利用质子转移反应飞行时间质谱仪(PTR-ToF-MS, 奥地利Ionicon公司)测定, 时间分辨率为5 min.
2 结果与讨论 2.1 老化时间对SOA生成潜势的影响环境空气在OFR内经·OH氧化之后质量增加的部分, 即相对于Bypass反应器增加的部分即为二次颗粒物生成量, 其中OA增加的部分即为SOA生成(SOA生成量=OAOFR-OABypass). 实验结果表明, OFR中的90%以上的二次颗粒物生成为SOA, 这可能和OFR高浓度的·OH环境以及气相反应对二次无机颗粒物的生成贡献有限有关.
老化时间是影响SOA生成量的重要因素. 在外场观测实验中, 为了最大化OFR内SOA生成量, 测得SOA生成潜势, 需要确定和选择外场条件下SOA生成的最佳老化时间. 在观测之前, 通过预实验测量了外场条件下北京大气在不同老化时间下的SOA生成量, 如图 2所示. 从中可以看出, SOA生成量随老化时间的增加先增加后减少. 在SOA生成量增加阶段, 主要以加氧反应为主, 含氧官能团逐渐增加; 在生成量减少阶段, 过长的老化时间会使颗粒相有机物碳碳键发生断裂, 从而形成小分子强挥发性物质进入到气相中[18, 29], 使得颗粒相质量损失. 从而存在最佳的老化时间使得SOA生成量最大, 最大的SOA生成量即为SOA的生成潜势. SOA生成量的变化除了来自前体物VOCs的氧化, 还可能受OA继续氧化的影响.然而, 根据OFR实验中AMS测量的OA粒径谱, SOA生成主要来源于小于100 nm的新生成颗粒物的贡献, 已有的OA在OFR中的继续氧化对其质量的影响可以忽略[30].因此, 在本实验条件下可以忽略OA的继续老化对SOA生成潜势的影响, 只考虑前体物VOCs对SOA生成潜势的贡献.

|
图 2 北京大气SOA生成量与老化时间的关系 Fig. 2 Relationship between SOA formation and aging time in urban Beijing |
根据实验结果, 当老化时间达到12 d左右时, 北京大气的SOA生成量达到最大. 而在洛杉矶城市大气中利用OFR开展外场实验的结果显示, 当老化时间在0.8~6 d时SOA生成量最大[23], 其他外场环境中进行的OFR实验也显示当SOA生成量最大时老化时间可能在2~3 d[31]、0.4~1.5 d[30]等范围内变化. 北京大气SOA生成最大时的老化时间明显高于发达国家, 这可能是由于不同环境大气中的VOCs组成及浓度水平存在差异, 将在另一篇文章中进行详细讨论. 根据测量结果, 为保证SOA的生成量达到最大, 本实验中将OFR内的老化时间控制在12 d左右.
2.2 SOA生成潜势的时间变化规律北京大气SOA生成量随测量时间变化明显. 图 3(a)显示了7月26~28日的一段测量结果, 从中可以看出SOA生成潜势在7月27日04:00和7月28日0:00左右出现两个明显峰值, 呈现夜间高白天低的变化趋势, 这与Ortega等[23]报道的结果一致. 图 3(b)显示OA也呈现夜间高白天低的趋势, 其中一个原因可能是由于大气边界层厚度在夜间压缩变薄使污染物聚集, 因此导致了夜间污染物的水平高于白天[32], 这将在后面进行更详细地讨论.

|
图 3 典型污染物随时间的变化趋势 Fig. 3 Trends of typical pollutants over time |
通过分析2017年5月1日至9月30日期间的全部外场数据, 统计了整个夏天SOA生成潜势的日变化规律, 如图 4(a)所示. 结果显示夏季SOA生成潜势均值在3.9~9.4 μg·m-3范围内变化, 并且呈现夜间高白天低的趋势. Ortega等[23]在洛杉矶进行的OFR实验结果也显示相同的SOA生成潜势昼夜变化规律, 由于洛杉矶特殊的气象条件, 其认为洛杉矶SOA生成潜势的昼夜变化可能主要受传输控制. 北京夏季的昼夜风向无明显变化规律, SOA生成潜势可能主要受大气VOCs的氧化程度的影响. 为排除边界层高度变化对SOA生成潜势的影响, 本研究使用一氧化碳(CO)作为背景浓度示踪物[33], 用SOA生成潜势与CO的比值表征SOA生成潜势的变化, 如图 4(b)所示. 结果显示排除边界层高度变化的影响后, SOA生成潜势仍然显示出夜间高白天低的趋势, 这表明除了边界层高度变化这个因素外还有其他的因素影响SOA生成潜势.

|
箱形图的5个特征点从上到下分别表示95%、75%、中位数、25%、5%的数值点, 中间实心方点表示数据的平均值 图 4 SOA生成潜势日变化规律分析 Fig. 4 Diurnal variation analysis of SOA formation potential |
在16:00左右, SOA生成潜势达到最低点(~3.9 μg·m-3), 同时伴随O3浓度达到峰值, 如图 5(a)所示, 表明SOA生成潜势可能受光化学反应的影响. 受强烈光化学反应的影响, 这个阶段环境大气中VOCs的消耗达到最大值, 进入OFR的VOCs量相应地达到最低值, 这些VOCs经氧化后生成的SOA也因此达到低值. 所以, 图 4(a)中出现的SOA潜势最低点可能是由于前体物VOCs浓度降低导致生成的SOA减少.

|
甲苯浓度由PTR-ToF-MS测定,臭氧浓度来自于奥体中心国控点(2017年夏季) 图 5 环境中甲苯、苯和臭氧的日变化规律 Fig. 5 Diurnal variation analysis of benzene, toluene, and ozone |
苯和甲苯是人为源排放的典型代表VOCs, 甲苯作为SOA的重要前体物, 对SOA的生成潜势有重要影响. 观测期间, 苯和甲苯的日变化规律如图 5(b)所示, 浓度分别在1.99~3.48 μg·m-3和1.54~3.09 μg·m-3之间变化, 属于Liu等[34]报道的清洁天气与轻度污染天的浓度范围, 与夏季北京多以清洁天气为主的情况一致[35]. 苯和甲苯浓度都在07:00左右达到峰值, 16:00左右达到最低点. 北京机动车排放对总VOCs的贡献达36%~48%[34, 36], 交通高峰造成的机动车排放增加, 将使得VOCs的浓度日变化会出现双峰分布[37]. 本实验中苯和甲苯在夜间没有出现峰值, 可能由于夜间也受边界层压缩的影响, 导致交通晚高峰时段苯和甲苯浓度总体呈上升趋势. 如图 5(c)所示, VOCs的浓度与CO的比值的日变化则显示19:00左右出现不明显的峰值, 说明VOCs的日变化的确受边界层高度变化的影响. 苯和甲苯午后出现的浓度低值则是由于夏季午后日照变强, 强烈的光化学反应使得VOCs消耗增加[37, 38], 这个结果与SOA生成潜势的减少相一致. 另外, SOA生成潜势与环境大气中甲苯浓度的相关关系如图 6(a)所示, 相关系数 r2 为0.80, SOA生成潜势与OFR中甲苯的消耗量相关关系如图 6(b)所示, r2 =0.69, 都呈现较好的相关性. 这些结果充分说明午后SOA生成潜势的降低是因为夏季强烈的光化学反应使得VOCs消耗量增加, 从而使得SOA潜势随VOCs浓度的变化密切相关.

|
图 6 SOA生成潜势与环境大气甲苯浓度、甲苯消耗量的相关关系 Fig. 6 Correlation between SOA formation potential and the concentration of ambient toluene or the consumption of ambient toluene |
Palm等[30]在以单萜烯为主要VOCs的针叶林中进行的OFR实验结果显示, SOA生成潜势均值在夜间最高为1.3μg·m-3, 白天则最高为0.2 μg·m-3. Ortega等[23]在洛杉矶城市大气中进行的OFR实验显示, SOA生成潜势均值在夜间最高为4.0 μg·m-3, 白天则最高为2.0 μg·m-3. 相比北京城市大气来说, 这些地方的SOA生成潜势要小得多. 从影响SOA生成的角度分析, VOCs起重要作用. 比如, 针叶林中VOCs主要是以植物排放的高活性低浓度VOCs为主[30], 这些VOCs在大气中容易被氧化而生成SOA, 进而使其SOA生成潜势降低. 与人为源排放相比, 这些VOCs的浓度也比较低, 能够贡献的SOA生成潜势也低. 同样是城市大气, 北京城市大气的VOCs组成与洛杉矶接近, 而在浓度上北京城市大气中VOCs比洛杉矶要高很多[39~42], 这可能也是北京地区SOA生成潜势高的重要原因. 北京环境大气的高SOA生成潜势, 可能对北京地区细颗粒物污染具有重要贡献.
3 结论(1) 基于OFR估算的北京夏季城市大气SOA生成潜势具有明显的昼夜变化特征, 除了边界层变化之外, 受白天强烈的光化学反应的影响, SOA生成潜势显示夜间高白天低的趋势. 在较强大气氧化性导致环境大气VOCs消耗的情况下, SOA生成潜势在16:00左右达到最低.
(2) 通过与国外的SOA生成潜势对比, 北京环境大气由于VOCs浓度高, 其SOA生成潜势明显高于发达国家清洁地区, 可能对北京地区细颗粒物污染具有重要贡献.
| [1] | Huang R J, Zhang Y L, Bozzetti C, et al. High secondary aerosol contribution to particulate pollution during haze events in China[J]. Nature, 2014, 514(7521): 218-222. DOI:10.1038/nature13774 |
| [2] |
杜博涵, 黄晓锋, 何凌燕, 等. 宁波市PM2.5中碳组分的时空分布特征和二次有机碳估算[J]. 环境科学, 2015, 36(9): 3128-3134. Du B H, Huang X F, He L Y, et al. Seasonal and spatial variations of carbon fractions in PM2.5 in ningbo and the estimation of secondary organic carbon[J]. Environmental Science, 2015, 36(9): 3128-3134. |
| [3] |
曹军骥, 李建军. 二次有机气溶胶的形成及其毒理效应[J]. 地球环境学报, 2016, 7(5): 431-441. Cao J J, Li J J. Formation and toxicological effect of secondary organic aerosols[J]. Journal of Earth Environment, 2016, 7(5): 431-441. DOI:10.7515/JEE201605001 |
| [4] | Hallquist M, Wenger J C, Baltensperger U, et al. The formation, properties and impact of secondary organic aerosol:current and emerging issues[J]. Atmospheric Chemistry and Physics:Discussions, 2009, 9(14): 5155-5236. DOI:10.5194/acp-9-5155-2009 |
| [5] | Jimenez J L, Canagaratna M R, Donahue N M, et al. Evolution of organic aerosols in the atmosphere[J]. Science, 2009, 326(5959): 1525-1529. DOI:10.1126/science.1180353 |
| [6] | Tkacik D S, Presto A A, Donahue N M, et al. Secondary organic aerosol formation from intermediate-volatility organic compounds:Cyclic, linear, and branched alkanes[J]. Environmental Science & Technology, 2012, 46(16): 8773-8781. |
| [7] | Goldstein A H, Galbally I E. Known and unexplored organic constituents in the Earth's atmosphere[J]. Environmental Science & Technology, 2007, 41(5): 1514-1521. |
| [8] |
谢绍东, 田晓雪. 挥发性和半挥发性有机物向二次有机气溶胶转化的机制[J]. 化学进展, 2010, 22(4): 727-733. Xie S D, Tian X X. Formation mechanism of secondary organic aerosols from the reaction of volatile and semi-volatile compounds[J]. Progress in Chemistry, 2010, 22(4): 727-733. |
| [9] |
郭松, 胡敏, 尚冬杰, 等. 基于外场观测的大气二次有机气溶胶研究[J]. 化学学报, 2014, 72(2): 145-157. Guo S, Hu M, Shang D J, et al. Research on secondary organic aerosols basing on field measurement[J]. Acta Chimica Sinica, 2014, 72(2): 145-157. |
| [10] | Kang E, Root M J, Toohey D W, et al. Introducing the concept of potential aerosol mass (PAM)[J]. Atmospheric Chemistry and Physics:Discussions, 2007, 7(22): 5727-5744. DOI:10.5194/acp-7-5727-2007 |
| [11] |
王倩, 陈长虹, 王红丽, 等. 上海市秋季大气VOCs对二次有机气溶胶的生成贡献及来源研究[J]. 环境科学, 2013, 34(2): 424-433. Wang Q, Chen C H, Wang H L, et al. Forming potential of secondary organic aerosols and sources apportionment of VOCs in autumn of Shanghai, China[J]. Environmental Science, 2013, 34(2): 424-433. |
| [12] |
吕子峰, 郝吉明, 段菁春, 等. 北京市夏季二次有机气溶胶生成潜势的估算[J]. 环境科学, 2009, 30(4): 969-975. Lü Z F, Hao J M, Duan J C, et al. Estimate of the formation potential of secondary organic aerosol in Beijing summertime[J]. Environmental Science, 2009, 30(4): 969-975. |
| [13] | Odum J R, Hoffmann T, Bowman F, et al. Gas/particle partitioning and secondary organic aerosol yields[J]. Environmental Science & Technology, 1996, 30(8): 2580-2585. |
| [14] |
陈文泰, 邵敏, 袁斌, 等. 大气中挥发性有机物(VOCs)对二次有机气溶胶(SOA)生成贡献的参数化估算[J]. 环境科学学报, 2013, 33(1): 163-172. Chen W T, Shao M, Yuan B, et al. Parameterization of contribution to secondary organic aerosol (SOA) formation from ambient volatile organic compounds (VOCs)[J]. Acta Scientiae Circumstantiae, 2013, 33(1): 163-172. |
| [15] | Donahue N M, Robinson A L, Stanier C O, et al. Coupled partitioning, dilution, and chemical aging of semivolatile organics[J]. Environmental Science & Technology, 2006, 40(8): 2635-2643. |
| [16] | Donahue N M, Epstein S A, Pandis S N, et al. A two-dimensional volatility basis set:1.Organic-aerosol mixing thermodynamics[J]. Atmospheric Chemistry and Physics:Discussions, 2011, 11(7): 3303-3318. DOI:10.5194/acp-11-3303-2011 |
| [17] | Kang E, Toohey D W, Brune W H. Dependence of SOA oxidation on organic aerosol mass concentration and OH exposure:experimental PAM chamber studies[J]. Atmospheric Chemistry and Physics:Discussions, 2011, 11(4): 1837-1852. DOI:10.5194/acp-11-1837-2011 |
| [18] | Lambe A T, Onasch T B, Croasdale D R, et al. Transitions from functionalization to fragmentation reactions of laboratory secondary organic aerosol (SOA) generated from the OH oxidation of alkane precursors[J]. Environmental Science & Technology, 2012, 46(10): 5430-5437. |
| [19] | Hall W A, Pennington M R, Johnston M V. Molecular transformations accompanying the aging of laboratory secondary organic aerosol[J]. Environmental Science & Technology, 2013, 47(5): 2230-2237. |
| [20] | Lambe A T, Cappa C D, Massoli P, et al. Relationship between oxidation level and optical properties of secondary organic aerosol[J]. Environmental Science & Technology, 2013, 47(12): 6349-6357. |
| [21] | Ahlberg E, Falk J, Eriksson A, et al. Secondary organic aerosol from VOC mixtures in an oxidation flow reactor[J]. Atmospheric Environment, 2017, 161: 210-220. DOI:10.1016/j.atmosenv.2017.05.005 |
| [22] | Chu B W, Liu Y C, Ma Q X, et al. Distinct potential aerosol masses under different scenarios of transport at a suburban site of beijing[J]. Journal of Environmental Sciences, 2016, 39: 52-61. DOI:10.1016/j.jes.2015.11.003 |
| [23] | Ortega A M, Hayes P L, Peng Z, et al. Real-time measurements of secondary organic aerosol formation and aging from ambient air in an oxidation flow reactor in the Los Angeles area[J]. Atmospheric Chemistry and Physics:Discussions, 2016, 16(11): 7411-7433. DOI:10.5194/acp-16-7411-2016 |
| [24] | Hu W W, Hu M, Hu W, et al. Chemical composition, sources, and aging process of submicron aerosols in Beijing:contrast between summer and winter[J]. Journal of Geophysical Research:Atmospheres, 2016, 121(4): 1955-1977. DOI:10.1002/2015JD024020 |
| [25] | Li R, Palm B B, Ortega A M, et al. Modeling the radical chemistry in an oxidation flow reactor:radical formation and recycling, sensitivities, and the OH exposure estimation equation[J]. The Journal of Physical Chemistry A, 2015, 119(19): 4418-4432. DOI:10.1021/jp509534k |
| [26] | Mao J, Ren X, Brune W H, et al. Airborne measurement of OH reactivity during INTEX-B[J]. Atmospheric Chemistry and Physics:Discussions, 2009, 9(1): 163-173. DOI:10.5194/acp-9-163-2009 |
| [27] | Drewnick F, Hings S S, DeCarlo P, et al. A new time-of-flight aerosol mass spectrometer (TOF-AMS)-instrument description and first field deployment[J]. Aerosol Science and Technology, 2005, 39(7): 637-658. DOI:10.1080/02786820500182040 |
| [28] | Middlebrook A M, Bahreini R, Jimenez J L, et al. Evaluation of composition-dependent collection efficiencies for the aerodyne aerosol mass spectrometer using field data[J]. Aerosol Science and Technology, 2012, 46(3): 258-271. DOI:10.1080/02786826.2011.620041 |
| [29] | Lambe A T, Chhabra P S, Onasch T B, et al. Effect of oxidant concentration, exposure time, and seed particles on secondary organic aerosol chemical composition and yield[J]. Atmospheric Chemistry and Physics:Discussions, 2015, 15(6): 3063-3075. DOI:10.5194/acp-15-3063-2015 |
| [30] | Palm B B, Campuzano-Jost P, Ortega A M, et al. In situ secondary organic aerosol formation from ambient pine forest air using an oxidation flow reactor[J]. Atmospheric Chemistry and Physics:Discussions, 2016, 16(5): 2943-2970. DOI:10.5194/acp-16-2943-2016 |
| [31] | Tkacik D S, Lambe A T, Jathar S, et al. Secondary organic aerosol formation from in-use motor vehicle emissions using a potential aerosol mass reactor[J]. Environmental Science & Technology, 2014, 48(19): 11235-11242. |
| [32] | Han S Q, Bian H, Tie X X, et al. Impact of nocturnal planetary boundary layer on urban air pollutants:Measurements from a 250-m tower over Tianjin, China[J]. Journal of Hazardous Materials, 2009, 162(1): 264-269. DOI:10.1016/j.jhazmat.2008.05.056 |
| [33] | De Gouw J, Jimenez J L. Organic aerosols in the earth's atmosphere[J]. Environmental Science & Technology, 2009, 43(20): 7614-7618. |
| [34] | Liu C T, Ma Z B, Mu Y J, et al. The levels, variation characteristics, and sources of atmospheric non-methane hydrocarbon compounds during wintertime in Beijing, China[J]. Atmospheric Chemistry and Physics:Discussions, 2017, 17(17): 10633-10649. DOI:10.5194/acp-17-10633-2017 |
| [35] |
谢志英, 刘浩, 唐新明, 等. 北京市近12年空气污染变化特征及其与气象要素的相关性分析[J]. 环境工程学报, 2015, 9(9): 4471-4478. Xie Z Y, Liu H, Tang X M, et al. Air pollution variation characteristics and its correlation with meteorological data in Beijing in recent 12 years[J]. Chinese Journal of Environmental Engineering, 2015, 9(9): 4471-4478. DOI:10.12030/j.cjee.20150960 |
| [36] | Wu R R, Li J, Hao Y F, et al. Evolution process and sources of ambient volatile organic compounds during a severe haze event in Beijing, China[J]. Science of the Total Environment, 2016, 560-561: 62-72. DOI:10.1016/j.scitotenv.2016.04.030 |
| [37] |
张靖, 邵敏, 苏芳. 北京市大气中挥发性有机物的组成特征[J]. 环境科学研究, 2004, 17(5): 1-5. Zhang J, Shao M, Su F. Study on composition of ambient volatile organic compounds (VOCs) in Beijing city[J]. Research of Environmental Sciences, 2004, 17(5): 1-5. |
| [38] |
张俊刚, 王跃思, 吴方堃, 等. 北京市大气中BTEX工作日与非工作日的浓度变化[J]. 环境化学, 2009, 28(1): 112-116. Zhang J G, Wang Y S, Wu F K, et al. Difference between workday and non-workday of BTEX concentration in Beijing urban area[J]. Environmental Chemistry, 2009, 28(1): 112-116. |
| [39] | Li J, Wu R, Li Y, et al. Effects of rigorous emission controls on reducing ambient volatile organic compounds in Beijing, China[J]. Science of the Total Environment, 2016, 557-558: 531-541. DOI:10.1016/j.scitotenv.2016.03.140 |
| [40] | Duan J C, Tan J H, Yang L, et al. Concentration, sources and ozone formation potential of volatile organic compounds (VOCs) during ozone episode in Beijing[J]. Atmospheric Research, 2008, 88(1): 25-35. DOI:10.1016/j.atmosres.2007.09.004 |
| [41] | Pang Y B, Fuentes M, Rieger P. Trends in selected ambient volatile organic compound (VOC) concentrations and a comparison to mobile source emission trends in California's South Coast Air Basin[J]. Atmospheric Environment, 2015, 122: 686-695. DOI:10.1016/j.atmosenv.2015.10.016 |
| [42] | Baker A K, Beyersdorf A J, Doezema L A, et al. Measurements of nonmethane hydrocarbons in 28 United States cities[J]. Atmospheric Environment, 2008, 42(1): 170-182. DOI:10.1016/j.atmosenv.2007.09.007 |