 2017, Vol. 38
2017, Vol. 38

2. 浙江大学医学院附属邵逸夫医院, 杭州 310058;
3. 浙江大学公共卫生学院毒理学研究室, 杭州 310058;
4. 浙江大学公共卫生实验教学中心, 杭州 310058;
5. 浙江大学药学院生化药学研究室, 杭州 310058
 , DING Rui1 , HUANG Dan-ni1 , ZHU Zi-yi2 , ZHANG Jun3 , YE Huai-zhuang4 , XU Ying-chun5 , JIN Yong-tang1
, DING Rui1 , HUANG Dan-ni1 , ZHU Zi-yi2 , ZHANG Jun3 , YE Huai-zhuang4 , XU Ying-chun5 , JIN Yong-tang1
2. Department of Cardiothoracic Surgery, Sir Run Run Shaw Hospital, School of Medicine, Zhejiang University, Hangzhou 310058, China;
3. Department of Toxicology, School of Medicine, Zhejiang University, Hangzhou 310058, China;
4. Public Health Experimental Teaching Center, Zhejiang University, Hangzhou 310058, China;
5. School of Pharmacology, Zhejiang University, Hangzhou 310058, China
汽车尾气中含有大量有害物质, 包括由碳黑、焦油及重金属等构成的颗粒物(particulate matter, PM), 苯及苯系物, 多环芳烃(polycyclic aromatic hydrocarbon, PAH)以及NO2、CO、SO2及O3等有害气体[1].颗粒物可能通过引起系统或局部氧化应激、炎性反应, 导致人群呼吸道感染、哮喘恶化、癌症风险增加[2].苯是国际癌症研究中心认定的人类致癌物[3].另外, 迄今已发现200多种PAHs, 其中大部分具有致癌性.随着机动车数量的不断增长, 机动车尾气已经成为城市空气污染的主要来源[4, 5].
肺癌是目前最常见的原发性恶性肿瘤之一, 《2015年中国肿瘤登记年报》显示肺癌是我国目前死亡率最高的恶性肿瘤[6].多数流行病学研究表明, 空气污染与肺癌患者死亡率及住院率升高相关[7~9].随着肿瘤研究的深入, 人们逐渐认识到, 环境致癌物不仅可通过突变等遗传学机制改变基因的表达, 也可通过表观遗传学机制改变基因功能, 导致肺癌发生[10, 11]. DNA甲基化是目前表观遗传学研究中最为清楚, 也是最重要的现象[12].近年来, 围绕空气污染相关疾病的表观遗传学机制研究越来越多, 但仅限于单一空气污染物实验室暴露[13]和工厂工人职业暴露[14~16], 国内外关于交通污染现场暴露的研究较为少见.
p53基因是抑癌基因的一种, 其主要功能是促进损伤细胞凋亡[17].有研究表明, p53基因低甲基化与DNA双键断裂以及染色体不稳定相关[18], 并参与了肺癌的发生发展[19].有研究发现, 工厂工人空气污染物PAHs及PM10职业暴露会引起p53基因低甲基化[20~22]. MGMT(O6-methylguanine DNA methyltranferase)基因的主要功能是通过移除烷化剂加和物, 保护细胞免受烷化剂的致癌作用[23, 24].研究在肺癌等多种癌症细胞中都发现了MGMT基因高甲基化现象[25~27].有报道指出, 煤烟暴露引起的肺癌与MGMT基因高甲基化相关[28]. MAGE-I类抗原基因(melanoma-associated antigen gene-I)大多定位在X染色体上, 主要功能是促进肿瘤细胞的增殖转移, 一般只在睾丸和胎盘组织中表达[29]. MAGEs基因高表达在肺癌细胞中常见, 与肺癌发生发展相关[30~32].
综上所述, 环境污染物暴露可能通过引起p53、MGMT、MAGEs基因甲基化异常, 导致肺癌的发生.本研究分析了交通污染暴露对p53、MGMT、MAGE-A4基因甲基化的影响, 从表观遗传学角度探讨交通污染暴露的致病和致癌机制.
1 材料与方法 1.1 主要试剂及仪器仪器:中流量总悬颗粒物采样器(ZC-Q0101, 浙江恒达仪器仪表有限公司)、焦磷酸测序仪(PyroMark Q24, 美国Qiagen公司).试剂:组织基因组DNA提取试剂盒购自天根生物科技(北京)有限公司, 重亚硫酸盐纯化试剂盒、PCR扩增试剂盒、焦磷酸测序试剂盒均购自美国Qiagen公司.
1.2 实验动物与现场暴露60只清洁级健康Wistar大鼠(8周龄), 由浙江大学动物研究中心提供.大鼠购入后在动物房内适应一周, 饲养温度为20~24℃, 保持12 h/12 h昼夜交替, 可自由饮水进食.适应性喂养一周后, 按随机数字表法随机分5组, 每组6只.其中3组分别在隧道(高暴露组)、路口(中暴露组)、校园(对照组)暴露7 d, 另外2组分别在隧道暴露14 d/28 d, 暴露时间为每天07:30~19:30.暴露实验分别在春季、秋季各进行一次.本研究中的春季和秋季根据气象学季节的定义确定, 当日平均气温连续5 d高于10℃时为春季开始, 当日平均气温连续5 d低于22℃时为秋季开始.
1.3 环境检测采用中流量总悬浮颗粒物采样器测量暴露现场PM10的浓度, 每次自07:30~19:30, 每小时采样一次, 连续检测7 d, 污染物水平为84次测量值的平均值. NO2浓度的测定采用盐酸萘乙二胺比色法(Saltzman法).
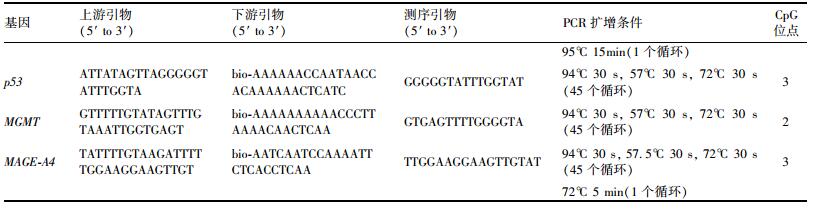
1.4 DNA甲基化检测暴露结束后, 将大鼠带回实验室, 称重, 经CO2吸入使其麻醉, 经左股动脉采血, -80℃保存.经颈椎脱臼处死大鼠, 解剖后, 经心脏用生理盐水进行灌注, 直至冲洗出的液体无血液.完整获取其肺组织, 用冷生理盐水冲洗后, 拭干、称重, -80℃保存.肺组织/血液DNA的提取按照DNA提取试剂盒所提供的方法进行. PCR和焦磷酸测序部分见文献[33], 引物序列和扩增条件详见表 1. PCR体系(25 μL):dNTPs和酶混合液12.5 μL, Coraload Concentrate 2.5 μL, 10 μmol·L-1引物各0.5 μL, 去离子水8 μL, 纯化DNA 1 μL. CpG位点甲基化百分率的取值范围为0~100%, 计算各检测CpG位点甲基化百分率的平均值, 将该值作为基因启动子区的甲基化水平.
|
|
表 1 DNA甲基化检测引物序列 Table 1 Primers and PCR conditions for DNA methylation analyses |
1.5 统计分析
采用SPSS 20.0统计软件对实验数据进行分析. DNA甲基化水平比较符合正态分布, 用平均值±标准差表示.根据各暴露现场污染物的浓度, 将隧道归为高暴露组, 路口归为中暴露组, 校园归为对照组.采用单因素方差法分析不同暴露组间DNA甲基化水平的差异, 采用Spearman相关分析法分析污染物与DNA甲基化的关系.所有统计学检验均采用双侧检验, 以P<0.05为差异有统计学意义.
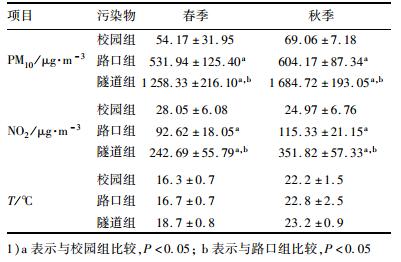
2 结果与分析 2.1 基本情况整个暴露过程中, 大鼠活动正常, 无器质性病变.暴露现场污染物浓度详见表 2, 主要污染物(PM10、NO2)浓度均为隧道(高暴露组)>路口(中暴露组)>校园(对照组), 差异具有统计学意义.另外, 秋季污染物浓度高于春季, 但差异无统计学意义.
|
|
表 2 春秋季各暴露地点主要交通相关空气污染物水平 Table 2 Mean concentrations of traffic-related air pollutants during the exposures |
2.2 暴露7 d后DNA甲基化水平比较
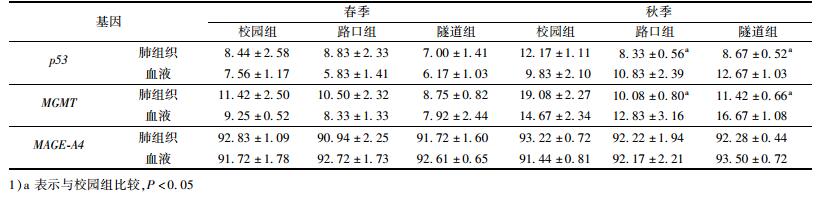
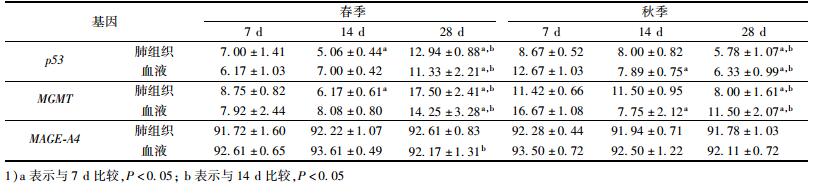
春季暴露7 d后, 在肺组织和血液中, 未发现p53、MGMT和MAGE-A4启动子甲基化水平在三组间存在显著的统计学差异.秋季暴露7 d后, 路口和隧道暴露组肺组织p53(P路口=0.000; P隧道=0.000) 和MGMT(P路口=0.000; P隧道=0.001) 启动子甲基化水平均低于校园组, 差异具有统计学意义, 而血液p53和MGMT启动子甲基化水平在各暴露组间均不存在统计学差异. MAGE-A4启动子区处于高度甲基化状态, 暴露7 d后, 在肺组织和血液中, 均未发现MAGE-A4启动子甲基化水平在三组间存在显著的统计学差异.详见表 3.
|
|
表 3 暴露7 d后大鼠肺组织/血液DNA甲基化水平1) Table 3 Promoter methylation changes measured after 7 d exposure |
2.3 DNA甲基化水平与污染物相关性分析
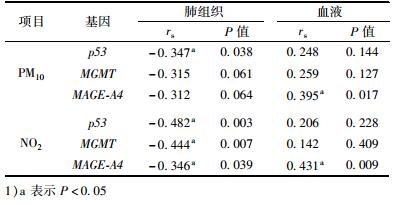
如表 4所示, 在肺组织中, PM10和p53启动子甲基化水平呈负相关关系(r=-0.347; P=0.038), 而与MGMT、MAGE-A4启动子甲基化水平的相关性无统计学意义; NO2和p53、MGMT、MAGE-A4启动子甲基化水平均存在负相关(r值分别为-0.482、-0.444、-0.346, P值均<0.05).在血液中, MAGE-A4启动子甲基化水平与PM10、NO2均呈正相关(r值分别为0.395、0.431, P<0.05); PM10、NO2与p53、MGMT启动子甲基化水平的相关性均无统计学差异.
|
|
表 4 DNA甲基化水平与污染物(PM10、NO2)的相关性分析结果(r值)1) Table 4 Association of PM10/NO2 average exposure with promoter methylation levels |
2.4 DNA甲基化水平与暴露时间的关系
利用隧道暴露组, 探讨在高浓度污染物暴露情况下, DNA甲基化水平与暴露时间的关联.如表 5所示, 春季暴露组中, 肺组织p53和MGMT启动子甲基化水平随暴露时间的增加先降低后升高; 隧道暴露28 d后, 血液p53和MGMT启动子甲基化水平开始升高.秋季暴露组中, 肺组织、血液p53和MGMT启动子甲基化水平随暴露时间的增加而逐渐降低.无论春季还是秋季, MAGE-A4启动子甲基化水平基本不随暴露时间增加而发生显著的改变.
|
|
表 5 隧道暴露7 d/14 d/28 d后大鼠肺组织/血液DNA甲基化水平比较 Table 5 Promoter methylation changes measured after 7 d/14 d/28 d exposures in the tunnel |
3 讨论
大量实验研究表明, 表观遗传学修饰异常与疾病的发生发展密切相关, 但目前仍不清楚表观遗传学修饰异常是疾病发生的原因还是结果[34, 35].本研究首次探索了交通污染暴露对p53、MGMT、MAGE-A4基因启动子甲基化水平的影响, 对揭示表观遗传修饰异常与疾病发生的因果关系具有重要的科学意义.
目前已知p53基因是细胞凋亡的一种正向调节因子; MGMT基因作为肿瘤抑制基因, 具有通过移除烷化剂加和物, 保护细胞免受烷化剂致癌作用的功能; MAGEs基因主要参与肿瘤细胞的增殖转移.有报道称, 肺癌病人细胞中存在p53、MGMT、MAGEs基因甲基化异常现象.本研究发现, 秋季隧道暴露7 d后, 肺组织中p53启动子甲基化水平显著降低, 而且随着暴露时间的增加, 甲基化水平进一步降低, 这与国内外其他研究结果相一致.有研究在暴露于高浓度PM和PAHs的工厂工人血液中发现了p53基因启动子低甲基化现象[21, 22]. Peluso等[15]对来自泰国某工厂工人、工厂附近居民及郊区居民的血样进行检测后发现, p53基因启动子甲基化水平随着居住地与工厂间距离的减小而降低.
有报道指出, 煤烟暴露会引起MGMT启动子高甲基化[28], 也有研究在肺癌细胞中检测到了MGMT高甲基化现象[26, 27], 与本研究发现的交通污染暴露会导致MGMT启动子低甲基化结果不一致.可能由于MGMT启动子低甲基化是空气污染暴露引起肺癌的一个早期事件, 也可能与不同地区、不同时期污染物水平, 污染物主要来源及研究对象不同有关, 需在今后的研究中进一步阐明.
本研究在肺癌病人的唾液中检测到了MAGE-A1和MAGE-B2启动子低甲基化现象[30]. MAGEs低甲基化是多种肿瘤早期筛检的指标之一, 包括肺癌[32, 36].但本研究发现交通污染暴露并不会导致MAGE-A4启动子甲基化水平发生显著改变.可能与MAGEs基因家族本身高度甲基化, 只在睾丸和胎盘组织中表达[29]相关, 也可能是MAGEs基因低甲基化是肿瘤发生后的结果.
有报道称[37], 季节是引起表观遗传改变的因素之一, 因此本研究分别在春季和秋季进行了一次现场暴露实验, 并比较了两阶段DNA甲基化水平的差异.笔者研究发现, 秋季对照组p53和MGMT启动子甲基化水平高于春季对照组, 可能与气温有关, 原因有待进一步研究.本研究还发现, 虽然各污染物水平在春秋两季的差异无统计学意义, 但表观遗传改变却存在季节差异.秋季暴露7 d后, 肺组织中p53、MGMT启动子甲基化水平显著降低, 而春季暴露7 d后, 未发现3组间甲基化水平存在统计学差异.另外, 在春季, 随着暴露时间的增加, 肺组织p53、MGMT启动子甲基化水平先降低后升高; 在秋季, 肺组织p53、MGMT启动子甲基化水平随着暴露时间的增加而逐渐降低.颗粒物是个混合物[38], 春秋结果不完全相同可能与颗粒物的组成成分不同有关, 也可能与湿度和温度的修饰效应[39]有关, 其原因有待进一步研究.
围绕空气污染相关疾病的表观遗传学机制研究多以外周血为研究样本.本研究首次比较了相同暴露条件下, 同一个体血液和肺组织样本中DNA甲基化水平改变模式的差异.秋季暴露7 d后, 路口和隧道暴露组肺组织p53、MGMT启动子甲基化水平与校园组的差异就有统计学意义, 而并没有发现血液中这两个基因启动子甲基化水平在3组之间存在统计学差异.本研究选取了与交通污染最相关的PM10、NO2两种污染物[40]进行分析, Spearman相关分析结果也显示PM10、NO2的7 d暴露对肺组织中DNA甲基化水平的影响更大.这说明影响血液DNA甲基化的因素非常复杂, 除上述提到的因素外, 空气污染暴露引起的炎性反应、氧化应激等因素可能也起到一定作用[2, 41].但随着暴露时间的增加, 肺组织和血液中DNA甲基化水平改变模式趋于一致.上述结果表明, 血液样本易采集且能较好地反映肺组织中基因甲基化水平改变情况, 在实际工作中可以血液代表组织样本研究污染物暴露对组织基因甲基化水平的影响.同时建议在研究短期暴露效应时, 如果选择血液样本, 给出结果时要稍加注意.
本研究采用焦磷酸测序法检测基因甲基化水平, 其结果重复性强, 可以同时检测多个CpG位点的甲基化水平, 精确率更高.本次研究首次比较了肺组织和血液中DNA甲基化水平改变的差异.本文是现场暴露研究, 能综合评价各种因素(污染物、温度、噪声等)对DNA甲基化水平的影响, 但是难以确定各个因素的贡献大小.其次, 本研究以大鼠为实验对象, 上述结果能否在人群中重现仍需进一步验证.最后, 本研究以表观遗传学修饰异常改变为结局指标, 秋季隧道暴露28d后, p53和MGMT启动子甲基化水平与春季对照组差异并不大, 因此无法确定表观遗传学修饰异常改变与疾病发生相关.
4 结论(1) 交通污染暴露会导致p53和MGMT启动子低甲基化, 且DNA甲基化水平改变存在季节差异和组织差异, 秋季暴露更容易引起DNA甲基化水平发生改变, 肺组织中DNA甲基化水平改变比血液中更容易发生.
(2) 交通污染暴露会引起大鼠血液和肺组织中p53、MGMT启动子甲基化水平异常改变, 但表观遗传修饰异常改变后的结果还需进一步地研究.从表观遗传角度解释复杂疾病的发生机制[42, 43], 表观遗传学的药物研究和开发已成为肿瘤治疗研究的热点领域[44], 深入进行表观遗传效应研究, 不仅对揭示污染物的作用机制具有重要科学意义, 而且为空气污染所致疾病的防治提供了一个重要靶向.
| [1] | Heck J E, Wu J, Lombardi C, et al. Childhood cancer and traffic-related air pollution exposure in pregnancy and early life[J]. Environmental Health Perspectives, 2013, 121(11-12): 1385-1391. |
| [2] | Esposito S, Tenconi R, Lelii M, et al. Possible molecular mechanisms linking air pollution and asthma in children[J]. BMC Pulmonary Medicine, 2014, 14(1): 31. DOI:10.1186/1471-2466-14-31 |
| [3] |
叶丛雷, 谢品华, 秦敏, 等. 广州市交通主干道空气中苯系物的测量[J]. 环境科学, 2012, 33(11): 3718-3724. Ye C L, Xie P H, Qin M, et al. BTX monitoring nearby main road traffic in Guangzhou[J]. Environmental Science, 2012, 33(11): 3718-3724. |
| [4] | 孟夏, 陈仁杰, 阚海东. 我国交通来源大气污染现状及其健康危害[J]. 中华预防医学杂志, 2011, 45(11): 1043-1045. DOI:10.3760/cma.j.issn.0253-9624.2011.11.020 |
| [5] | Patel M M, Miller R L. Air pollution and childhood asthma: recent advances and future directions[J]. Current Opinion in Pediatrics, 2009, 21(2): 235-242. DOI:10.1097/MOP.0b013e3283267726 |
| [6] | Chen W Q, Zheng R S, Baade P D, et al. Cancer statistics in China, 2015[J]. CA: A Cancer Journal for Clinicians, 2016, 66(2): 115-132. DOI:10.3322/caac.21338 |
| [7] | Jardim M J. MicroRNAs: implications for air pollution research[J]. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, 2011, 717(1-2): 38-45. DOI:10.1016/j.mrfmmm.2011.03.014 |
| [8] | Holloway J W, Savarimuthu Francis S, Fong K M, et al. Genomics and the respiratory effects of air pollution exposure[J]. Respirology, 2012, 17(4): 590-600. DOI:10.1111/res.2012.17.issue-4 |
| [9] | Reid B C, Ghazarian A A, DeMarini D M, et al. Research opportunities for cancer associated with indoor air pollution from solid-fuel combustion[J]. Environmental Health Perspectives, 2012, 120(11): 1495-1498. DOI:10.1289/ehp.1204962 |
| [10] | Soberanes S, Gonzalez A, Urich D, et al. Particulate matter air pollution induces hypermethylation of the p16 promoter via a mitochondrial ROS-JNK-DNMT1 pathway[J]. Scientific Reports, 2012, 2: 275. |
| [11] | Bowman R V, Wright C M, Davidson M R, et al. Epigenomic targets for the treatment of respiratory disease[J]. Expert Opinion on Therapeutic Targets, 2009, 13(6): 625-640. DOI:10.1517/14728220902926119 |
| [12] | Yang I V, Schwartz D A. Epigenetic control of gene expression in the lung[J]. American Journal of Respiratory and Critical Care Medicine, 2011, 183(10): 1295-1301. DOI:10.1164/rccm.201010-1579PP |
| [13] | Rager J E, Smeester L, Jaspers I, et al. Epigenetic changes induced by air toxics: formaldehyde exposure alters miRNA expression profiles in human lung cells[J]. Environmental Health Perspectives, 2011, 119(4): 494-500. |
| [14] | Bollati V, Marinelli B, Apostoli P, et al. Exposure to metal-rich particulate matter modifies the expression of candidate microRNAs in peripheral blood leukocytes[J]. Environmental Health Perspectives, 2010, 118(6): 763-768. DOI:10.1289/ehp.0901300 |
| [15] | Peluso M, Bollati V, Munnia A, et al. DNA methylation differences in exposed workers and nearby residents of the Ma Ta Phut industrial estate, Rayong, Thailand[J]. International Journal of Epidemiology, 2013, 41(6): 1753-1760. |
| [16] | Bollati V, Baccarelli A, Hou L F, et al. Changes in DNA methylation patterns in subjects exposed to low-dose benzene[J]. Cancer Research, 2007, 67(3): 876-880. DOI:10.1158/0008-5472.CAN-06-2995 |
| [17] | Robles A I, Linke S P, Harris C C. The p53 network in lung carcinogenesis[J]. Oncogene, 2002, 21(45): 6898-6907. DOI:10.1038/sj.onc.1205563 |
| [18] | Pavanello S, Pesatori A C, Dioni L, et al. Shorter telomere length in peripheral blood lymphocytes of workers exposed to polycyclic aromatic hydrocarbons[J]. Carcinogenesis, 2010, 31(2): 216-221. DOI:10.1093/carcin/bgp278 |
| [19] | Woodson K, Mason J, Choi S W, et al. Hypomethylation of p53 in peripheral blood DNA is associated with the development of lung cancer[J]. Cancer Epidemiology, Biomarkers & Prevention, 2001, 10(1): 69-74. |
| [20] | Pavanello S, Bollati V, Pesatori A C, et al. Global and gene-specific promoter methylation changes are related to anti-B[J]. International Journal of Cancer, 2009, 125(7): 1692-1697. DOI:10.1002/ijc.v125:7 |
| [21] | Alegría-Torres J A, Barretta F, Batres-Esquivel L E, et al. Epigenetic markers of exposure to polycyclic aromatic hydrocarbons in Mexican brickmakers: a pilot study[J]. Chemosphere, 2013, 91(4): 475-480. DOI:10.1016/j.chemosphere.2012.11.077 |
| [22] | Hou L F, Zhang X, Tarantini L, et al. Ambient PM exposure and DNA methylation in tumor suppressor genes: a cross-sectional study[J]. Particle and Fiber Toxicology, 2011, 8(1): 25. DOI:10.1186/1743-8977-8-25 |
| [23] | Fahrer J, Kaina B. O6-methylguanine-DNA methyltransferase in the defense against N-nitroso compounds and colorectal cancer[J]. Carcinogenesis, 2013, 34(11): 2435-2442. DOI:10.1093/carcin/bgt275 |
| [24] | Palmisano W A, Divine K K, Saccomanno G, et al. Predicting lung cancer by detecting aberrant promoter methylation in sputum[J]. Cancer Research, 2000, 60(21): 5954-5958. |
| [25] | Esteller M, Hamilton S R, Burger P C, et al. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia[J]. Cancer Research, 1999, 59(4): 793-797. |
| [26] | Zöchbauer-Müller S, Fong K M, Virmani A K, et al. Aberrant promoter methylation of multiple genes in non-small cell lung cancers[J]. Cancer Research, 2001, 61(1): 249-255. |
| [27] | Bujko M, Kowalewska M, Danska-Bidzinska A, et al. The promoter methylation and expression of the O6-methylguanine-DNA methyltransferase gene in uterine sarcoma and carcinosarcoma[J]. Oncology Letters, 2012, 4(3): 551-555. |
| [28] | Liu Y, Lan Q, Shen M, et al. Aberrant gene promoter methylation in sputum from individuals exposed to smoky coal emissions[J]. Anticancer Research, 2008, 28(4B): 2061-2066. |
| [29] | 徐莹莹, 桑梅香, 单保恩. 肿瘤抗原MAGE与p53关系的研究进展[J]. 癌变·畸变·突变, 2013, 25(2): 152-154. |
| [30] | Olaussen K A, Soria J C, Park Y W, et al. Assessing abnormal gene promoter methylation in paraffin-embedded sputum from patients with NSCLC[J]. European Journal of Cancer, 2005, 41(14): 2112-2119. DOI:10.1016/j.ejca.2005.06.013 |
| [31] | Gure A O, Chua R, Williamson B, et al. Cancer-testis genes are coordinately expressed and are markers of poor outcome in non-small cell lung cancer[J]. Clinical Cancer Research, 2005, 11(22): 8055-8062. DOI:10.1158/1078-0432.CCR-05-1203 |
| [32] | Grunwald C, Koslowski M, Arsiray T, et al. Expression of multiple epigenetically regulated cancer/germline genes in nonsmall cell lung cancer[J]. International Journal of Cancer, 2006, 118(10): 2522-2528. DOI:10.1002/(ISSN)1097-0215 |
| [33] | Ding R, Jin Y T, Liu X N, et al. Characteristics of DNA methylation changes induced by traffic-related air pollution[J]. Mutation Research/Genetic Toxicology and Environmental Mutagenesis, 2016, 796: 46-53. DOI:10.1016/j.mrgentox.2015.12.002 |
| [34] | Cortessis V K, Thomas D C, Levine A J, et al. Environmental epigenetics: prospects for studying epigenetic mediation of exposure-response relationships[J]. Human Genetics, 2012, 131(10): 1565-1589. DOI:10.1007/s00439-012-1189-8 |
| [35] | Lovinsky-Desir S, Miller R L. Epigenetics, asthma, and allergic diseases: a review of the latest advancements[J]. Current Allergy and Asthma Reports, 2012, 12(3): 211-220. DOI:10.1007/s11882-012-0257-4 |
| [36] | Liu F, Killian J K, Yang M, et al. Epigenomic alterations and gene expression profiles in respiratory epithelia exposed to cigarette smoke condensate[J]. Oncogene, 2010, 29(25): 3650-3664. DOI:10.1038/onc.2010.129 |
| [37] | Ricceri F, Trevisan M, Fiano V, et al. Seasonality modifies methylation profiles in healthy people[J]. PLoS One, 2014, 9(9): e106846. DOI:10.1371/journal.pone.0106846 |
| [38] | Hou L F, Zhang X, Zheng Y N, et al. Altered methylation in tandem repeat element and elemental component levels in inhalable air particles[J]. Environmental and Molecular Mutagenesis, 2014, 55(3): 256-265. DOI:10.1002/em.21829 |
| [39] |
罗娜娜, 赵文吉, 晏星, 等. 交通与气象因子对不同粒径大气颗粒物的影响机制研究[J]. 环境科学, 2013, 34(10): 3741-3748. Luo N N, Zhao W J, Yan X, et al. Study on influence of traffic and meteorological factors on inhalable particle matters of different size[J]. Environmental Science, 2013, 34(10): 3741-3748. |
| [40] | Kelly F J, Fussell J C. Air pollution and airway disease[J]. Clinical & Experimental Allergy, 2011, 41(8): 1059-1071. |
| [41] | Sørensen M, Daneshvar B, Hansen M, et al. Personal PM2.5 exposure and markers of oxidative stress in blood[J]. Environmental Health Perspectives, 2003, 111(2): 161-166. |
| [42] | Karmaus W, Ziyab A H, Everson T, et al. Epigenetic mechanisms and models in the origins of asthma[J]. Current Opinion in Allergy and Clinical Immunology, 2013, 13(1): 63-69. DOI:10.1097/ACI.0b013e32835ad0e7 |
| [43] | Fuso A. The 'golden age' of DNA methylation in neurodegenerative diseases[J]. Clinical Chemistry and Laboratory Medicine, 2013, 51(3): 523-534. |
| [44] | Hatzimichael E, Crook T. Cancer epigenetics: new therapies and new challenges[J]. Journal of Drug Delivery, 2013: 529312. |