 2017, Vol. 38
2017, Vol. 38



2. 中国水产科学研究院淡水渔业研究中心, 农业部淡水渔业和种质资源利用重点实验室, 无锡 214081
2. Key Laboratory of Genetic Breeding and Aquaculture Biology of Freshwater Fishes, Ministry of Agriculture, Freshwater Fisheries Research Center, Chinese Academy of Fishery Sciences, Wuxi 214081, China
水资源循环利用在当今时代成为研究热点, 高效的水体处理方法成为此热点的关键[1, 2].目前, 人工湿地系统在工业、生活和农业废水处理过程中都得到广泛应用[3].中国作为世界上的水产大国, 在水产养殖过程中, 多数养殖池塘都属于封闭性水域, 由于缺乏科学性管理并且盲目增加养殖密度和投喂量等增加了养殖水体负荷, 易导致水体中N、P等营养物含量过高和病菌滋生, 若不进行有效地处理就直接排放到外界, 会对湖泊水体和土壤等带来二次污染.因此, 人工湿地系统应用于渔业生产过程中, 对于保障水产品质量安全和外界环境优化等方面具有重要意义.
人工湿地系统是由植物, 滤料和微生物群落三大部分组成[4].微生物群落附着于滤料外表面及其孔隙和植物根系表层进行生长[5].滤料种类和植物根系分泌的氧气、小分子有机酸、维生素、促生长激素等能够影响根际区微生物的生长和群落的组成.因此, 微生物、滤料、植物三者之间存在一定的关联性, 并且三者都能够影响人工湿地系统的净化功能.相关研究表明, 微生物能够降解吸收氮化物, 磷化物, 有机物和其他营养物, 对于水质优化具有明显效果[6].为深入分析微生物对人工湿地系统净化功能的作用, 先进的微生物研究方法必不可少, 高通量测序技术具有通量高、平行测序和成本低廉等优势, 能够成为微生物研究的重要手段, 相关研究结果也证实了这一点[7~11].
湿地系统中不同植物种类的根系分泌物组成成分具有一定的差异, 这对于根际微生物的数量和物种组成具有十分重要的影响[12, 13].研究表明, 不同植物和不同滤料构建湿地系统的净水效率具有明显的差异性[14, 15].根据查阅相关资料, 目前所研究的湿地植物种类繁多, 但同时具有净水功能和经济价值, 又体现区域特色的水生植物较少; 所研究的滤料大多数都是常规应用的人工湿地滤料.因此, 本研究选用慈菇和茭白作为人工湿地植物, 考虑到慈菇和茭白是长江下游常见的水生经济植物并且根系较发达易富集微生物; 选用石榴石和磁铁矿作为人工湿地滤料, 考虑到石榴石和磁铁矿都是化学稳定性好的新型耐磨净水材料并且孔隙度较高易截留微生物, 另外, 磁铁矿能够富集有害微生物[16].为了提高湿地系统净化养殖水体的效率并分析系统中根系微生物群落的多样性, 本研究通过在湿地模型正常运行的情况下, 比较分析不同模型中的根际微生物群落结构特征和多样性特性, 旨在为湿地植物的优化选择和植物滤料搭配的最佳模型提供一定的理论支持和技术指导.
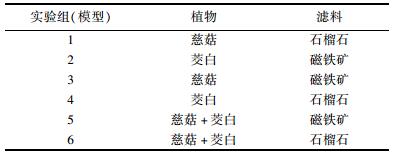
1 材料与方法 1.1 湿地模型的构建和样品采集小型湿地模型构建的人工湿地植物是茭白和慈菇, 滤料是石榴石和磁铁矿.根据多变量实验设计方法进行设计, 分别对两种植物和两种滤料进行组合, 共6种组合.如表 1所示.
|
|
表 1 每个湿地模型的植物和滤料组成 Table 1 Plants and filter material compositions of the six wetland microcosm combinations |
湿地模型主要是由进水口、出水口、滤料、植物和塑料桶组成(图 1).湿地模型的基本数据如表 2所示, 其中水力负荷和水力停留时间计算详见公式(1) 和(2).所有的湿地模型建立在养殖池塘旁(图 2), 实验地点在中国水产科学研究院淡水渔业研究中心大浦实验基地.小型湿地模型根据表 1中的植物和滤料组成, 其中研究的茭白植株相互之间株型相似, 慈菇植株相互之间株型相似, 每个模型栽种健康无病的植株4株, 滤料填埋深度为0.550 m.利用进水口的阀门控制水的流速, 保持所有实验组合水体流速相同.进水口连接养殖池塘, 池塘养殖模式为主养鲫鱼, 兼养鲢鱼、鳙鱼、草鱼和青鱼, 是长江下游典型的池塘养殖模式.

|
图 1 小型湿地模型示意 Fig. 1 Diagrammatic drawing of a wetland microcosm |
|
|
表 2 小型湿地模型基本数据 Table 2 Basic data of the wetland microcosms |


|
图 2 小型湿地模型实物 Fig. 2 Photograph of the wetland microcosms |
湿地模型构建运行后, 每隔10 d采样一次, 在上午10:00~12:00采样, 采集每一个湿地模型的植物根系于6号自封袋中, 采集后将样品放入4℃冰箱待测, 并于3 d之内提取所有样品的DNA.
1.3 根系微生物DNA的提取预处理:植物根系和滤料样品采集后, 将其置于灭菌后的锥形瓶中, 加入200 mL无菌水振荡, 采用恒温摇床在225 r·min-1振速下振荡30 min, 然后静置5 min, 取悬浮液10 mL在10 000 r·min-1下离心10 min, 弃上清, 重复两次, 收集沉淀, 保存于-20℃冰箱中用于DNA提取.
细菌DNA提取:提取方法参照Zhou等[17]的方法, 沉淀样品中加入270 μL DNA提取缓冲液(0.1 mol·L-1 PBS缓冲液pH=8, 0.1mol·L-1 EDTA pH=8, 0.1 mol·L-1 Tris pH=8, 1.5 mol·L-1 NaCl, 1% CTAB和5 μL蛋白酶K(10 mg·mL-1), 于37℃下225 r·min-1摇30 min, 加入30 μL的20% SDS, 65℃水浴2 h, 于室温10 000 r·min-1离心10 min, 取上清液; 沉淀中加入90 μL DNA提取缓冲液, 10 μL 20% SDS, 漩涡振荡10 s, 65℃水浴10 min, 离心10 min; 取上清液, 重复以上步骤3次, 将3次上清均匀混合, 与等体积的氯仿/异戊醇混合, 10 000 r·min-1离心10 min, 吸取上层水相加入0.6倍体积的异丙醇混匀, 10 000 r·min-1离心10 min; 沉淀用70%的冰乙醇洗涤, 晾干, 加入50 μL TE溶解, -20℃保存.
1.4 PCR扩增及产物定量和均一化本研究采用引物515F 5′-GTGCCAGCMGCC GCGG-3′和907R 5′-CCGTCAATTCMTTTRAGTTT-3′的细菌16S rDNA V4-V5区部分序列[18].整个实验过程中所有涉及的PCR扩增都是采用20 μL的PCR反应体系, 体系中各组分及用量如下:4 μL的FastPfu Buffer(5×), 2 μL的dNTPs(2.5 mmol·L-1), 0.8 μL Forward Primer(5 μmol·L-1), 0.8 μL的Reverse Primer(5μmol·L-1), 0.4 μL的FastPfu Polymerase, 10 ng的Template DNA, 然后补充ddH2O至20 μL.使用PCR扩增仪ABI GeneAmp® 9700型进行扩增反应.扩增程序为:95℃预变性2 min; 95℃变性30 s, 55℃退火30 s, 72℃延伸30 s, 25个循环; 72℃延伸5 min.全部样品按照正式试验条件进行, 每个样品3个重复, 将同一样品的PCR产物混合后用2%琼脂糖凝胶电泳检测, 使用AxyPrepDNA凝胶回收试剂盒(AXYGEN公司)切胶回收PCR产物, Tris_HCl洗脱; 2%琼脂糖电泳检测.参照电泳初步定量结果, 将PCR产物用QuantiFluorTM-ST蓝色荧光定量系统(Promega公司)进行检测定量, 之后按照每个样品的测序量要求, 进行相应比例的混合.
1.5 PE(Pair-end)文库构建及高通量测序将产物连接“Y”字形接头, 使用磁珠筛选去除接头自连片段, 利用PCR扩增进行文库模板的富集, 采用氢氧化钠变性后得到单链DNA片段.应用高通量测序平台Illumina MiSeq进行高通量测序, 具体过程如下:DNA片段的一端与引物碱基互补, 固定在芯片上; 另一端随机与附近的另外一个引物互补, 也被固定住, 形成“桥”; PCR扩增, 产生DNA簇; DNA扩增子线性化成为单链; 加入改造过的DNA聚合酶和带有4种荧光标记的dNTP, 每次循环只合成一个碱基; 用激光扫描反应板表面, 读取每条模板序列第一轮反应所聚合上去的核苷酸种类; 将“荧光基团”和“终止基团”化学切割, 恢复3′端黏性, 继续聚合第二个核苷酸; 统计每轮收集到的荧光信号结果, 获知模板DNA片段的序列, 以上工作由上海凌恩生物科技有限公司协助完成.
高通量测序之后进行相关数据去杂优化后, 采用UPARSE(version 7.1 http://drive5.com/uparse/)在相似度97%水平下对优化序列进行OTU(operational taxonomic unit)聚类分析和分类学分析; 基于OTU进行多种多样性指数分析, 基于OTU聚类分析结果, 对OTU进行多种多样性指数分析以及测序深度的检测; 基于分类学信息, 可以在各个分类水平上进行群落结构的统计分析.在上述分析的基础上, 可以进行一系列群落结构和系统发育等深入的统计学和可视化分析.
2 结果与分析 2.1 各采集样品的微生物多样性本实验从湿地模型中共采集30个样本用于根系微生物的研究.根据物种累积曲线显示(图 3), 本实验中所采集样品足以反映出此次微生物的丰富度[19].并且所有样品的OTU序列的相似性为97%, 样品中OTU的数量范围为1 191~2 884.代表微生物群落丰富度的Ace和Chao指数分别为2 279~3 777和1 807~3 734.

|
图 3 物种累积曲线 Fig. 3 Species accumulation curve |
Shannon-Wiener指数是研究群落物种数及个体数和分布均匀程度的综合指标[20], 能够表明微生物群落的多样性.本研究中, Shannon指数在4.22~6.37之间, Shannon指数在2号09-18的样本中最高, 在1号08-18最低(表 3).另外, 不同湿地根系微生物的Shannon指数值在不同采样时间具有一定差异性.整个研究过程中, 湿地模型2号和4号的Shannon指数平均数值最高(分别为5.72和5.81), 1号和3号的Shannon指数平均数值最低(分别为5.23和5.35).
|
|
表 3 不同湿地模型在不同采样时间的Shannon指数 Table 3 Shannon indices of the six wetland microcosms at each sampling time |

通过高通量测序得出的OTUs共属52个门.其中, 丰富度较大的微生物属于变形菌门(Proteobacteria)和拟杆菌门(Bacteroidetes), 而较少的物种属于热袍菌门(Thermotogae), RsaHF231和SM2F11.变形菌门(Proteobacteria)在每一个样品中所占的比例最大, 最低能达到43.53%, 最高达70.45%.拟杆菌门(Bacteroidetes)在所有样品中所占的比例范围是10.84%~36.01%.另外, 在每个样品的所有的微生物物种中, β-变形菌(β-Proteobacteria)所占的比例最大(图 4).根据图 4, 湿地模型1和3中β-变形菌(β-Proteobacteria)的比例在所有湿地模型中最高, 湿地模型2和4中β-变形菌(β-Proteobacteria)的比例在所有湿地模型中最低, 湿地模型5和6中β-变形菌(β-Proteobacteria)的比例基本相等并处于前两者之间(图 4).其中, 慈菇和茭白根系微生物中变形菌门的平均比例分别为61.97%和51.78%.

|
图 4 每个样品中微生物物种的分布 Fig. 4 Distribution of microbial species in each sample |
通过样本在PCA图中的距离能够观察到样本之间的微生物多样性差异[21].每一个采集的根系样品的微生物群落结构特征具有一定的差异(图 5).其中, 当外界影响因素贡献率达到39.7%时, 8月18号的样品(红色圈), 8月28号的样品(蓝色圈)和9月8号、18号、28号的样品(黑色圈)在PC1轴上已明显地分开(图 5).

|
图 5 多样本PCA分析 Fig. 5 Multiple sample PCA |
为了深入分析不同湿地模型在不同采样时间的相似性, 本研究对测序结果进行了UPMGA聚类分析.结果表明, 8月份采集样品的根系微生物多样性和9月相比具有明显的差异(图 6).根据图 6, 每次采样中模型1号和3号微生物多样性高度相似, 而2号和4号高度相似, 5号和6号高度相似.另外, 1、4和6号在每次采样中根系微生物多样性具有很大的差异; 2、3和5号在每次采样中也具有类似的结果.

|
图 6 根际样品的UPGMA聚类分析 Fig. 6 UPMGA cluster analysis of rhizosphere samples |
相关研究表明, 在自然条件下, 根系微生物群落结构多样性的变化受植物分泌的根系分泌物和其他有机物质等影响[12, 13].根据Shannon指数分析并结合表 1, 在相同种植数量下, 种植茭白的湿地模型根系微生物多样性最高, 种植慈菇的最低, 而两种都种植的Shannon指数数值处于两者之间.此结果表明, 茭白的根系微环境能够更好地适合微生物的生存, 可能的原因是茭白根系分泌的小分子有机酸、维生素、促生长激素等分泌物比慈菇更加丰富与多样.另外, 同种处理模式的湿地模型随着运行, 其Shannon指数整体呈现先增加后平稳的趋势, 可能的原因是温度的变化、根系生长状况的影响和根际微环境中营养物质的变动等.
根据图 4和表 1, 慈菇的根系能够更好地富集β-变形菌(β-Proteobacteria), 而茭白能够更好地富集厌氧绳杆菌(Anaerolineae), 可能的原因是慈菇的根系微环境比茭白更适合β-变形菌(β-Proteobacteria)的生长和繁殖, 而茭白的根系环境更适合厌氧绳杆菌(Anaerolineae)的生长, 然而β-变形菌(β-Proteobacteria)含有一定数量的有害微生物物种, 因此茭白在富集有益微生物方面可能具有更强的能力.随着实验模型的运行, 所有模型中黄杆菌纲(Flavobacteria)和芽孢杆菌(Bacilli)比例逐渐下降, 而δ-变形菌(δ-Proteobacteria)、ε-变形菌(ε-Proteobacteria)、拟杆菌门(Bacteroidetes)、厌氧绳杆菌(Anaerolineae)和酸杆菌门(Acidobacteria)等多数微生物比例逐渐增加(图 4), 可能的原因是温度的变化、特定营养物质的变动和根系分泌物成分及数量的变化, 表明微生物的多样性逐渐增加, 此结果与Shannon指数相符合(表 3).
根据图 5和图 6, 不同湿地模型运行的不同时间段中根系微生物多样性始终处于变化中, 只有在运行后期9月的8、18和28号不同湿地模型差异性较小, 可能的原因是养殖水体对湿地模型的冲刷, 根系微环境稳态受到攻击需要一定时间的修复.结合表 1分析, 相同植物湿地模型的根系微生物多样性相似度高于相同滤料的湿地模型.可以推断出, 在本研究中, 两种植物对根系微生物富集都具有良好的能力, 而两种滤料表现则较弱, 可能的原因是滤料只是简单地为微生物提供附着场所, 而植物则为微生物提供了氧气、有机酸等必须的营养物质.
3.2 各湿地模型微生物群落多样性及其组成在整个研究期间, 植物根系微环境中涌现了大量的有益微生物.例如:玫瑰弯菌属(Roseiflexus sp.)在含氮污染物去除方面具有重要作用, 并且在缺氧的环境下具有异养硝化还原性能.硝化螺菌属(Nitrospira sp.)能将污染水体中的亚硝酸盐氧化成硝酸盐, 在水体净化氮循环中具有十分重要的作用[22].并且其丰富度能够提供氨氮, 亚硝酸盐和硝酸盐循环转化的效率, 能够保护养殖水体环境和避免因亚硝酸盐过多引起的鱼类死亡.存在于根系微环境中的小梨形菌属(Pirellula sp.)在氮循环中也具有十分重要的作用[23].硫杆菌属(Desulforhopalus sp.)在根系样品中被检测到, 其存在表明本研究中的湿地模型具有一定重金属去除的能力[24].根瘤菌属(Rhizobium sp.)和绿针假单胞菌(Pseudomonas chlororaphis)能够在根系微环境中通过分泌有机酸和磷酸酶将有机磷分解成无机磷(主要是H2PO4-和HPO42-)供植物吸收[25].红环菌科(Rhodocyclaceae sp.)在根系微环境中非常丰富, 能够在厌氧光照的环境下通过分解不同有机物质获得碳源进行生存, 在有机物质去除方面具有一定的作用[26]. Tepidicella sp.也大量存在于根系微环境中, 它能够在光照和厌氧的环境下通过分解有机物质、硫化物和氨氮生成氢供体和碳源进行生存繁殖. Dechloromonas sp.能够降低氯含量, 红假单胞菌属(Rhodopseudomonas sp.)能够吸收硫化氢.另外, Comamonadaceae sp.能够降解苯酚.除了有益微生物, 小部分的有害微生物也存在于各湿地模型的根系微环境中.例如, Desulfobulbusn sp.能够降解硫化物形成硫化氢, 对鱼体有一定的伤害.根瘤杆菌属(Rhizobacter sp.)是植物病原体, 而丹毒丝菌属(Erysipelothrix sp.)是对鱼体产生危害的病原体.
雷旭等对梭鱼草(Pontederia cordata)、美人蕉(Canna indica)和再力花(Thalia dealbata)湿地植物的根系微生物进行研究, 得到美人蕉根际能更好地富集周围的微生物并且具有提高微生物群落的多样性的能力[27].另外, 在相同种植数量的情况下, 不同植物种类对于根系微生物群落多样性具有一定的影响.有关研究表明, 利用PCR-DGGE技术对马铃薯(Solanum mberosum)、草莓(Fragari)和油菜(Brassicanapu)的根系微生物多样性进行分析研究, 三者之间的根系微生物群落结构具有很大的差异性[28].因此, 每种植物在微生物富集方面都具有不同的能力, 科研人员应该对不同的湿地植物进行选择, 筛选出具有净水效果最佳的湿地植物种类.由于技术的局限性和数据处理的复杂, 本研究仅对两种植物的根际微生物的功能菌群进行了相关研究.以后的研究工作应扩大湿地植物的优化选择和建立相关的湿地植物根系微生物文库, 以便于人工湿地的最佳构建和根系功能菌群的比较分析.
4 结论(1) 利用高通量测序技术, 实现了对茭白和慈菇根系微生物种类组成在不同模型中的直接测定, 初步探明两种植物在微生物富集方面的差异性.
(2) 茭白比慈菇在微生物富集方面具有更强的能力, 但慈菇能够更好地富集变形菌.
(3) 在本研究中, 两种植物对根系微生物富集都具有良好的能力, 其中, β-变形菌纲为实验期间不同植物根际富集的主要细菌类群, 而两种滤料表现则较弱.
(4) 根系微生物富含玫瑰弯菌属(Roseiflexus sp.)和硝化螺菌属(Nitrospira sp.)等大量的有益菌, 而如Desulfobulbusn sp.等有害微生物含量则较少.
| [1] | Smakhtin V, Revenga C, Döll P. A pilot global assessment of environmental water requirements and scarcity[J]. Water International, 2004, 29(3): 307-317. DOI:10.1080/02508060408691785 |
| [2] | Iasur-Kruh L, Hadar Y, Milstein D, et al. Microbial population and activity in wetland microcosms constructed for improving treated municipal wastewater[J]. Microbial Ecology, 2010, 59(4): 700-709. DOI:10.1007/s00248-009-9611-z |
| [3] | Vymazal J. The use constructed wetlands with horizontal sub-surface flow for various types of wastewater[J]. Ecological Engineering, 2009, 35(1): 1-17. DOI:10.1016/j.ecoleng.2008.08.016 |
| [4] | Greenway M. Suitability of macrophytes for nutrient removal from surface flow constructed wetlands receiving secondary treated sewage effluent in Queensland, Australia[J]. Water Science and Technology, 2003, 48(2): 121-128. |
| [5] | Watnick P, Kolter R. Biofilm, city of microbes[J]. Journal of Bacteriology, 2000, 182(10): 2675-2679. DOI:10.1128/JB.182.10.2675-2679.2000 |
| [6] | Faulwetter J L, Gagnon V, Sundberg C, et al. Microbial processes influencing performance of treatment wetlands: a review[J]. Ecological Engineering, 2009, 35(6): 987-1004. DOI:10.1016/j.ecoleng.2008.12.030 |
| [7] | Ligi T, Oopkaup K, Truu M, et al. Characterization of bacterial communities in soil and sediment of a created riverine wetland complex using high-throughput 16S rRNA amplicon sequencing[J]. Ecological Engineering, 2014, 72: 56-66. DOI:10.1016/j.ecoleng.2013.09.007 |
| [8] | Buée M, Reich M, Murat C, et al. 454 Pyrosequencing analyses of forest soils reveal an unexpectedly high fungal diversity[J]. New Phytologist, 2009, 184(2): 449-456. DOI:10.1111/j.1469-8137.2009.03003.x |
| [9] | Medinger R, Nolte V, Pandey R V, et al. Diversity in a hidden world: potential and limitation of next-generation sequencing for surveys of molecular diversity of eukaryotic microorganisms[J]. Molecular Ecology, 2010, 19(S1): 32-40. |
| [10] | Arfi Y, Buée M, Marchand C, et al. Multiple markers pyrosequencing reveals highly diverse and host-specific fungal communities on the mangrove trees Avicennia marina and Rhizophora stylosa[J]. FEMS Microbiology Ecology, 2012, 79(2): 433-444. DOI:10.1111/fem.2011.79.issue-2 |
| [11] | dos Santos H F, Cury J C, do Carmo F L, et al. Mangrove bacterial diversity and the impact of oil contamination revealed by pyrosequencing: bacterial proxies for oil pollution[J]. PLoS One, 2011, 6(3): e16943. DOI:10.1371/journal.pone.0016943 |
| [12] | Miethling R, Wieland G, Backhaus H, et al. Variation of microbial rhizosphere communities in response to crop species, soil origin, and inoculation with Sinorhizobium meliloti L33[J]. Microbial Ecology, 2000, 40(1): 43-56. DOI:10.1007/s002480000021 |
| [13] | Marschner P, Yang C H, Lieberei R, et al. Soil and plant specific effects on bacterial community composition in the rhizosphere[J]. Soil Biology and Biochemistry, 2001, 33(11): 1437-1445. DOI:10.1016/S0038-0717(01)00052-9 |
| [14] | Gersberg R M, Elkins B V, Lyon S R, et al. Role of aquatic plants in wastewater treatment by artificial wetlands[J]. Water Research, 1986, 20(3): 363-368. DOI:10.1016/0043-1354(86)90085-0 |
| [15] | Adcock P W, Ganf G G. Growth characteristics of three macrophyte species growing in a natural and constructed wetland system[J]. Water Science and Technology, 1994, 29(4): 95-102. |
| [16] | Li J H, Pan Y X. Environmental factors affect magnetite magnetosome synthesis in Magnetospirillum magneticum AMB-1: implications for biologically controlled mineralization[J]. Geomicrobiology Journal, 2012, 29(4): 362-373. DOI:10.1080/01490451.2011.565401 |
| [17] | Zhou J, Bruns M A, Tiedje J M. DNA recovery from soils of diverse composition[J]. Applied and Environmental Microbiology, 1996, 62(2): 316-322. |
| [18] | Xiong J B, Liu Y Q, Lin X G, et al. Geographic distance and pH drive bacterial distribution in alkaline lake sediments across Tibetan Plateau[J]. Environmental Microbiology, 2012, 14(9): 2457-2466. DOI:10.1111/emi.2012.14.issue-9 |
| [19] | Maughan H, Wang P W, Caballero J D, et al. Analysis of the cystic fibrosis lung microbiota via serial Illumina sequencing of bacterial 16S rRNA hypervariable regions[J]. PLoS One, 2012, 7(10): e45791. DOI:10.1371/journal.pone.0045791 |
| [20] | Müller A K, Westergaard K, Christensen S, et al. The diversity and function of soil microbial communities exposed to different disturbances[J]. Microbial Ecology, 2002, 44(1): 49-58. DOI:10.1007/s00248-001-0042-8 |
| [21] | Wang Y, Sheng H F, He Y, et al. comparison of the levels of bacterial diversity in freshwater, intertidal wetland, and marine sediments by using millions of illumina tags[J]. Applied and Environmental Microbiology, 2012, 78(23): 8264-8271. DOI:10.1128/AEM.01821-12 |
| [22] | Pester M, Maixner F, Berry D, et al. NxrB encoding the beta subunit of nitrite oxidoreductase as functional and phylogenetic marker for nitrite-oxidizing Nitrospira[J]. Environmental Microbiology, 2014, 16(10): 3055-3071. DOI:10.1111/emi.2014.16.issue-10 |
| [23] | Rabus R, Gade D, Helbig R, et al. Analysis of N-acetylglucosamine metabolism in the marine bacterium Pirellula sp. strain 1 by a proteomic approach[J]. Proteomics, 2002, 2(6): 649-655. DOI:10.1002/1615-9861(200206)2:6<>1.0.CO;2-O |
| [24] | Purdy K J, Nedwell D B, Embley T M. Analysis of the sulfate-reducing bacterial and methanogenic archaeal populations in contrasting Antarctic sediments[J]. Applied and Environmental Microbiology, 2003, 69(6): 3181-3191. DOI:10.1128/AEM.69.6.3181-3191.2003 |
| [25] | Chakrabarti J, Chatterjee S, Ghosh S, et al. Synergism of VAM and Rhizobium on production and metabolism of IAA in roots and root nodules of Vigna Mungo[J]. Current Microbiology, 2010, 61(3): 203-209. DOI:10.1007/s00284-010-9597-2 |
| [26] | Chung J, Shin S, Oh J. Characterization of a microbial community capable of reducing perchlorate and nitrate in high salinity[J]. Biotechnology Letters, 2009, 31(7): 959-966. DOI:10.1007/s10529-009-9960-1 |
| [27] |
雷旭, 李冰, 李晓, 等. 复合垂直流人工湿地系统中不同植物根际微生物群落结构[J]. 生态学杂志, 2015, 34(5): 1373-1381. Lei X, Li B, Li X, et al. Rhizosphere microbial communities of three plants in vertical-flow constructed wetland[J]. Chinese Journal of Ecology, 2015, 34(5): 1373-1381. |
| [28] | Smalla K, Wieland G, Buchner A, et al. Bulk and rhizosphere soil bacterial communities studied by denaturing gradient gel electrophoresis: plant-dependent enrichment and seasonal shifts revealed[J]. Applied and Environmental Microbiology, 2001, 67(10): 4742-4751. DOI:10.1128/AEM.67.10.4742-4751.2001 |