 2017, Vol. 38
2017, Vol. 38

 , ZHOU Jian-nan , LIU Bao-xian
, ZHOU Jian-nan , LIU Bao-xian , WANG Yan , ZHANG Bo-tao , SHI Ai-jun , YANG Dong-yan , CHANG Miao
, WANG Yan , ZHANG Bo-tao , SHI Ai-jun , YANG Dong-yan , CHANG Miao
近年来我国发生多次区域性大气重污染事件, 使公众更加关注大气细颗粒物PM2.5污染[1].在逆温、高湿等气象条件的作用下, PM2.5通过形成霾降低大气能见度、危害人体健康以及对区域或全球的气候变化产生影响[2, 3]. PM2.5中含有各种化学组分, 其中NH4+、NO3-、SO42-等水溶性离子对颗粒物质量有重要贡献.这些离子可来自一次排放源的排放, 但主要来自大气中气-粒转化过程形成的二次气溶胶.其气态的前体物主要有NH3、NOx(主要包括NO、NO2) 和SO2等酸碱性气体[4].
一些研究显示, 大气中的前体气体可以通过光化学反应形成大气颗粒物; 也可以通过成核或凝结核增长等物理过程形成大气颗粒物[5~9], 这些研究均指出大气中NOx和SO2被氧化成酸性气体HNO3和H2SO4, O3、H2O2和·OH等氧化剂对NOx和SO2转化成酸性气体起到重要的作用. SO2在大气中通常发生气相均相氧化、液相氧化以及在大气细颗粒物表面的非均相氧化; NOx主要以气相均相氧化为主.大气中酸性气体与碱性气体NH3发生反应, 生成NH4NO3、(NH4)2SO4和NH4HSO4, 或直接在酸性颗粒物表面反应形成NH4+、NO3-、HSO42-和SO42-. Hassan等[5]给出NH4+、NO3-和SO42-与各前体气体的反应机制, 但并未阐明气-粒浓度变化关系. Hsu等[6]指出SO42-和NH4+的浓度会随着SO2浓度的增加而增加; NO3-受NO2影响较小; NH4+与NH3相关性不大.李丽平等[7]总结NH4+和SO42-与其前体气体NH3和SO2之间有显著的相关关系, 但NO3-与前体气体HNO3之间的相关关系不显著. Zhuang等[8]研究表明各前体气体转化过程中受温湿度影响较大, 以及富氨地区大气PM2.5中NH4+浓度主要受SO2和NOx的浓度影响.朱彤等[9]的研究表明, 重污染事件过程中, 颗粒物表面的非均相转化对SO42-和NO3-的形成起到更为重要的作用.
以往北京地区有关PM2.5的研究多集中于化学组分的来源和分布特征, 对PM2.5中化学组分和前体气体关系的研究相对较少.实际上, 气-粒关系对大气中的酸碱气体和颗粒物中化学组分的寿命有重要影响[10], 也会影响大气中气态和颗粒态等化学组分的浓度分布特征.北京地区气候和地理条件与以往报道中的研究地区有很大区别, 分析北京地区PM2.5中SNA与前体气体的污染特征和相互关系一方面可以验证以往研究中的结果, 另一方面, 深入了解北京地区PM2.5的来源及控制因素, 有助于首都大气环境管理措施的制定和执行.
1 材料与方法 1.1 样品的采集采样点位于北京城区海淀区西二环与西三环之间, 监测中心7楼楼顶 (39°55′51″N, 116°19′9″E), 离车公庄西路约50 m.该位置属于北京市中心城区, 可在一定程度上代表北京市城区区域大气污染水平, 站点信息见图 1.本研究采样时段均为00:00~24:00, 采样时间为2015年1月1日~2015年12月31日, 使用Thermo2025i型单通道颗粒物采样器进行PM2.5样品的采集, 采样流量为16.7 L ·min-1, 采样膜采用whatman Φ 47 mm特氟龙材质滤膜.采样前将滤膜放入恒温恒湿箱中平衡24 h, 用梅特勒-XP6百万分之一精密电子天平称重后放入冰箱中低温保存.采样后将滤膜放入恒温恒湿箱平衡24 h, 称重后同样将滤膜样品低温保存在冰箱中等待分析.使用Thermo 42i NOx在线测量仪、Thermo 17i NH3在线测量仪、Thermo 43i SO2在线测量仪和Thermo 49i O3在线测量仪分别获得NOx、NH3、SO2和O3的实时数据; 4种污染气体均1 h收集一次数据, 所用分析数据为24 h日均值.共获得325套有效样品.

|
图 1 综合观测站点示意 Fig. 1 Location of the monitoring station |
本研究选取整张样品膜进行分析, 将滤膜放入一次性聚四氟乙烯消解管中, 超纯水定容至标线, 保持样品膜于水面下, 使用超声波清洗器提取1 h, 为防止含N组分的挥发及损失, 超声过程中放置一定数量的冰块.样品取出冷却至室温, 混匀后用一次性0.45 μm过滤头过滤后分析.实验用水均为ELGA-ULTRA仪器超滤至电阻率达18.2 MΩ ·cm的超纯水.水溶性无机阴离子NO3-、SO42-采用DionexICS-5000离子色谱仪进行分析; 水溶性阳离子NH4+使用DionexICS-2000离子色谱仪进行分析.阴、阳离子色谱柱型号分别为: AS11-HC和CS12A;阴离子淋洗液为30 mmol ·L-1 KOH溶液, 阳离子淋洗液为25 mmol ·L-1甲烷磺酸淋洗液, 流速均为1.0 mL ·min-1, 进样量均为50 μL.
1.3 数据质量和控制为保证气态前体物 (NOx、SO2、NH3和O3) 观测数据质量, 采用以下方法进行质量控制.
NOx和SO2测量仪每两天用标准钢瓶气进行零点和80%满量程的自动校准, 零点误差小于5%满量程, 80%点误差小于10%满量程. NO和NO2的检出限均是0.4×10-9, SO2的检出限是0.5×10-9.每个季度检查仪器线性和精密度, 斜率在0.99~1.01之间, 截距小于1%满量程, 相关系数大于0.999, 精密度20%点小于5×10-9, 80%点小于10-8.
NH3由于气体黏性大, 响应速度慢, 每个月校准一次NH3测量仪, 用标准钢瓶气进行零点和80%满量程的自动校准, 零点误差小于5%满量程, 80%点误差小于10%满量程. NH3检出限是10-9.每个季度检查仪器线性和精密度, 斜率在0.99~1.01之间, 截距小于1%满量程, 相关系数大于0.999, 精密度20%点小于5×10-9, 80%点小于10-8.
O3测量仪用经过计量传递的准确的臭氧发生器进行校准.每两天进行零点和80%满量程的自动校准, 零点误差小于5%满量程, 80%点误差小于10%.80%点误差不是满量程10%, 而是80%点本身的10%. O3检出限是1×10-9.每个季度检查仪器线性和精密度, 斜率在0.99~1.01之间, 截距小于1%满量程, 相关系数大于0.999, 精密度20%点小于5×10-9, 80%点小于10-8.
1.4 数据分析在线仪器测定的SO2、NH3、NOx和O3单位为10-9(体积分数), PM2.5中NH4+、NO3-、SO42-浓度单位为μg ·m-3, 分析时, 将SO2、NH3、NOx和O3的单位转化为μg ·m-3, 换算公式如下:
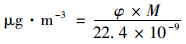
|
(1) |
式中, φ为各污染气体的体积分数;M为各污染气体的相对摩尔质量;22.4为该气体的摩尔体积, 即在标准状况[0℃(273K)、1.01×105 Pa]下, 1 mol任何理想气体所占的体积都约为22.4 L.
2 结果与讨论 2.1 北京大气中各前体气体及SNA的浓度水平大气中的NOx主要来自自然源和人为源排放.自然源排放主要包括微生物排放、闪电过程、平流层光化学过程和生物质自然燃烧等.人为源主要来自化石燃料燃烧和机动车尾气排放等[11].并且人为源排放高于自然源[12].2015年北京大气中NO的浓度范围为0.7~140 μg ·m-3, 平均值为17.7 μg ·m-3. NO2的浓度范围为10.1~147 μg ·m-3, 平均值为54.3 μg ·m-3, 高于环境空气质量二级标准NO2(40 μg ·m-3). NOx的平均值为72.0 μg ·m-3, 高于环境空气质量二级标准NOx(50 μg ·m-3)[13].同一站点2014年NO年平均值为22.2 μg ·m-3, NO2为57.9 μg ·m-3, 较去年相比, NO和NO2分别下降20.3%和6.2%.2015年北京大气中NO2浓度与王英等[14]观测到2011年北京、上海地区NO2浓度相当, 低于2009~2010年埃及开罗[5], 高于印度加尔各答[15]、2010年韩国首尔[16]、2006~2007年日本九州岛北岸[17], 具体数值见表 1.
|
|
表 1 各地大气中NO2、SO2和NH3的浓度比较/μg ·m-3 Table 1 Mass concentrations of NO2, SO2 and NH3at different sites/μg ·m-3 |
城市大气中的SO2主要来源于工业化石燃料燃烧和交通运输业[18].2015年北京大气中SO2的浓度范围为0.1~101 μg ·m-3, 平均浓度为14.2 μg ·m-3, 优于环境空气二级标准 (60 μg ·m-3)[13].同一站点2014年SO2年平均值为25.7 μg ·m-3, 较去年相比下降了44.7%.2015年北京大气中SO2的浓度低于2009~2010年埃及开罗[5]、2012年青岛[7]、2011年新加坡[10]、2011年北京、上海地区[14]、2004年秋季广州[18]和2006~2007年台湾[19], 高于2010年韩国首尔[16], 具体数值见表 1.
大气中的NH3主要来源于人类和牲畜的粪便、铵肥的挥发、生物质燃烧、农作物释放以及废水的排放等.在城市大气中, 机动车辆、污水处理过程、以及工业和燃烧过程也被认为是NH3的主要来源[20, 21].2015年北京大气中NH3的浓度范围为7.5~49.8 μg ·m-3, 平均浓度为21.5 μg ·m-3, 2015年北京大气中NH3的平均浓度高于2012年青岛[7]、2011年新加坡[10]、2010年韩国首尔[16]、2004年秋季广州[18]和2009年北京[22], 低于2009~2010年埃及开罗[5]、印度加尔各答[15]、2006~2007年台湾[19]和2005~2006年巴基斯坦拉合尔[23].具体数值见表 1.
大气细颗粒物PM2.5组分中, 无机盐离子占据最大部分, 质量浓度是PM2.5的40%~50%.而NO3-、SO42-和NH4+是PM2.5中最重要的无机盐, 占全部水溶性离子的80%~90%[7, 10, 17, 24, 27], 是表征区域污染的重要指标.观测期间, 3种主要水溶性离子NH4+、NO3-和SO42-浓度范围分别为0.0~64.3、0.1~112和0.4~100 μg ·m-3; 平均浓度分别为8.1、13.5和12.7 μg ·m-3, NO3->SO42->NH4+, 三者占PM2.5总量的43.4%.2014年同一站点观测到的NH4+、NO3-和SO42-年平均浓度分别10.4、17.6和15.0 μg ·m-3, 三者占PM2.5总量的51.1%.杨懂艳等[25]研究了2012~2013年北京城、郊NH4+、NO3-和SO42-年平均浓度分别13.5、20.3和19.4 μg ·m-3, 三者占PM2.5总量的59.2%, SNA无论在浓度水平还是PM2.5中的占比, 3年来均呈现下降的趋势.北京地区NH4+、NO3-和SO42-与广州、台湾、天津、西安地区相当; 高于青岛、新加坡、韩国首尔、日本九州岛北岸、上海、香港、纽约等地, 具体数值见表 2.
|
|
表 2 各地PM2.5中NH4+、SO42-和NO3-的浓度比较/μg ·m-3 Table 2 Mass concentrations of NH4+, SO42- and NO3- in PM2.5 at different sites/μg ·m-3 |
2.2 北京大气中各前体气体及SNA季节变化
北京大气中SO2平均浓度冬季最高, 为 (28.2±20.9) μg ·m-3, 春季和秋季次之, 夏季最低, 为 (5.0±5.0) μg ·m-3.从标准偏差来看, 表现出相同的规律, 浓度越高的季节, SO2的时间分布越不均匀, 见图 2.大气中SO2浓度在冬季最高, 一方面由于冬季为北京的采暖期, 大量的供热燃料使得SO2的排放源强明显高于其他季节, 且冬季逆温天气较多、大气层较稳定, 不利于大气中SO2的扩散稀释; 另一方面是冬季光化学反应较弱, SO2的氧化和转化能力降低[31].大气中SO2浓度在夏季最低, 最主要的原因是夏季燃煤较少, 同时温湿度高, 光化学作用强, 前体气体SO2容易转化成SO42-.另外, 夏季边界层较高, 对流强烈, 也有利于污染物的扩散和混匀.

|
图 2 2015年北京城区各前体气体及SNA浓度季节变化趋势 Fig. 2 Seasonal variation of concentrations of precursor gases and SNA in Beijing during 2015 |
北京大气中NO和NO2, 以平均浓度看, 冬季>秋季>春季>夏季, 冬季的平均浓度最高, 分别为 (39.3±37.7) μg ·m-3和 (65.1±31.9) μg ·m-3; 夏季平均浓度最低, 分别为 (2.50±1.7) μg ·m-3和 (42.2±9.4) μg ·m-3.从标准偏差来看, 同样表现出冬季最高, 夏季最低的特征, 即浓度越高的季节, NO和NO2的时间分布越不均匀, 这一点和SO2相同, 如图 2.郑晓霞等[11]的研究表明, 在自然源排放占主导的地区, NOx浓度一般在夏季出现最大值; 在人为源排放占主导的地区, NOx浓度一般在冬季出现最大值.由此可见, 北京市车公庄站点NOx污染排放始终是以人为源为主导, 即NOx主要受采暖季燃煤及机动车排放的影响.另一方面, 冬季逆温天气多、大气层较稳定, 不利于NOx的扩散稀释.而夏季空气对流运动强烈, 有利于NOx的扩散和混匀[5].
北京大气中NH3浓度特征不同于NOx和SO2, 以夏季最高, 为 (27.9±7.0) μg ·m-3, 春季次之, 为 (24.1±8.9) μg ·m-3, 冬季和秋季较低, 为 (17.8±6.3) μg ·m-3和 (16.4±4.7) μg ·m-3.从标准偏差来看, 春季最高, 夏季次之, 秋冬季较低, 说明虽然夏季浓度较高, 但与春季相比, 浓度分布较为均匀.秋冬季大气中NH3的浓度较低, 一方面由于温度较低、植被渐少, 不利于土壤和植被中NH3的排放; 另一方面是秋冬季温度低不利NH4NO3的分解; 而夏季及春季NH3的浓度较高, 一方面是受高温影响, NH3排放源变多变强, 另一方面高温有利于NH4NO3的分解, 导致NH3浓度升高[32~34].
北京市城区大气PM2.5中SNA质量浓度冬季>秋季>春季>夏季, 在PM2.5中百分含量 (体积分数) 分别秋季>夏季>春季>冬季.冬季SNA质量浓度最高, 但在PM2.5中占比最小, 是因为冬季采暖和静稳天气造成SNA浓度升高的同时OC、EC等其他组分也有所升高, 导致SNA占比下降.各组分变化见图 2和图 3, NH4+的浓度在冬季最高, 秋季次之, 春季和夏季较低.总体来讲, NH4+质量浓度全年变化并不明显.从在PM2.5中的百分含量看, NH4+在秋季占比最大, 冬季最小, 但四季相差不大, 维持在10%左右. NO3-的浓度以冬季最高, 其次为秋季和春季, 夏季最低.有研究指出[16, 17, 35, 36], NO3-浓度在春、冬季较高, 在夏、秋季较低, 与本研究的结果稍有不同, 这是因为2015年11月经历严重污染各污染物浓度飙升导致的.从百分含量看, NO3-在夏季占比较小, 为11.8%, 是由于NH4NO3在夏季高温条件下易挥发分解, NO3-浓度降低的结果. SO42-的浓度在冬季较高, 夏季和秋季次之, 春季最低.从百分含量看, SO42-在夏季占比最大, 为21.6%, 这是由于夏季SO42-在高温高湿光照强的环境下转化率增高, 且夏季PM2.5浓度较低双重作用的结果.

|
图 3 2015年北京城区PM2.5和SNA百分含量季节变化趋势 Fig. 3 Seasonal variation of concentration of PM2.5 and percentage of SNA in Beijing during 2015 |
有研究发现大气中NH3较少时, 首先与H2SO4反应生成NH4HSO4, NH3较多时NH4HSO4迅速转化成 (NH4)2SO4, NH3充足的环境下, NH3与H2SO4反应完全后, 进而与HNO3反应生成NH4NO3.当NH3不足时, 阴离子主要以HSO4-和SO42-形式存在[5, 10, 16].对NH4+和 (NO3-+2SO42-) 的离子浓度做散点图进行回归分析, 一元线性回归方程为y=0.97x, r=0.96;说明NH4+和 (NO3-+2SO42-) 两者之间显现出良好的相关性. ([NO3-]+2[SO42-])/[NH4+]比值小于1, 说明电荷基本平衡, 且NH4+过量, 即NH3能中和全部H2SO4和HNO3, 阴离子主要以SO42-和NO3-形式存在.从季节分布看, 春、夏、秋、冬季两者比值分别0.98、1.09、0.99和0.91, 见图 4.夏季 ([NO3-]+2[SO42-])/[NH4+]比值略大于1.0, 北京夏季NH3浓度最高, 说明不是NH3不足造成的, 是NO3-除与NH4+结合, 还可与Ca2+、Mg2+和Na+等阳离子结合的原因.

|
图 4 NH4+和 (NO3-+2SO42-) 平衡性分析 Fig. 4 Scatter plot of balance analysis between NH4+ and (NO3-+2SO42-) |
按照PM2.5质量浓度,优级0~35 μg ·m-3、良35~75 μg ·m-3、轻度污染75~115 μg ·m-3、中度污染115~150 μg ·m-3、重度污染150~250 μg ·m-3、严重污染>250 μg ·m-3,对PM2.5进行分级.研究不同空气质量级别下SO2、NO、NO2、NH3等前体气体和SNA的污染特征以及SNA对PM2.5质量浓度的贡献.
从优级天到严重污染天气, SO2呈上升趋势, 浓度范围为5.47~27.3 μg ·m-3.重度污染到严重污染, SO2有所下降, 是严重污染天气更适宜SO2在颗粒物表面发生非均相氧化反应造成的[37]; NO和NO2浓度范围分别为6.14~80.1 μg ·m-3和35.6~109 μg ·m-3; 分别升高了13倍和3倍; NH3整体变化不大, 并呈波动趋势, 浓度范围为17.0~26.6 μg ·m-3, 见图 5.

|
图 5 不同颗粒物污染等级下前体气体的质量浓度变化趋势 Fig. 5 Variation of concentrations of precursor gases at different PM2.5 levels |
随着PM2.5质量浓度的增加, NH4+、SO42-和NO3-这3种离子均有不同程度上升, NH4+从1.7 μg ·m-3增至34.2 μg ·m-3, NO3-从2.3 μg ·m-3增至52.9 μg ·m-3, SO42-从3.1 μg ·m-3增至43.3 μg ·m-3, 从数据看SO42-、NO3-积累均较迅速, 见图 6.比较3种离子在不同质量浓度级别下对PM2.5的贡献, NH4+的贡献率为8.7%~11.7%, 维持在相对稳定的水平. NO3-随着细颗粒质量级别的增加, 在PM2.5中的体积分数从11.7%增加到21.0%, 增幅率为79.5%.在重度污染时, 贡献率达到顶峰, 在严重污染时, 由于有机物等综合作用, 贡献率有所下降. SO42-的贡献率为14.1%~19.2%, 轻度污染时达到峰值, 随着污染级别升高, 有机物的急速攀升, SO42-的贡献率反而有所降低, 见图 7.随着污染程度的增加, NO3-/SO42-比值明显上升, 说明北京地区NO3-是重污染过程累积效应比较明显且贡献率最大的离子.

|
图 6 不同颗粒物污染等级下SNA的质量浓度变化趋势 Fig. 6 Variation of concentration of SNA at different PM2.5 levels |

|
图 7 不同颗粒物污染等级下SNA百分含量及NO3-/SO42-比值变化趋势 Fig. 7 Variation of percentage of SNA and ratio of NO3-/SO42- at different PM2.5 levels |
如图 5、图 6所示, 重污染事件过程中, 大气颗粒物、前体气体 (SO2和NOx) 质量浓度较高, 此时相对湿度也较高, 三者协同作用下前体气体在颗粒物表面发生非均相反应, 生成吸湿性很强的SO42-和NO3-, 使得颗粒物在大气RH下更容易吸收水, 从而显著增大其粒径与表面积浓度, 水的增加又进一步促进SO2和NO2等在颗粒物表面的非均相反应, 促使颗粒物吸湿增大, 以上这些过程形成一个正反馈机制, 可不断促进前体气体向二次气溶胶的转化.因此重污染中, 颗粒物表面的非均相转化对SO42-和NO3-的形成起到更为重要的作用[9].
2.5 北京大气中SNA及其前体气体的相关性分析利用SPSS软件分析北京大气PM2.5中NO3-、SO42-和NH4+与其前体气体的相关关系, 结果见表 3.除了NH3和SO2相关性较差, 其他气体之间均显著相关, NO、NO2和SO2之间相关系数>0.5, 这是由于这3种气体都有共同的供热燃煤排放源和局地工业排放源. NH3和NO2相关性较好, 是由于机动车尾气排放过程中同时排放NO2和NH3. SO42-、NO3-和NH4+等二次水溶性离子显著相关, 相关系数>0.8. NO3-与NO2、NO, NH4+与NH3, SO42-与SO2在置信度为0.01水平上均显著相关, 相关系数分别为0.68、0.47、0.46和0.39.
|
|
表 3 北京大气PM2.5中SNA及前体气体Spearman相关系数1) Table 3 Spearman correlations of precursor gases and SNA in PM2.5 in Beijing |
2.6 北京大气中SOR和NOR
由于NO3-和SO42-主要存在于粒径≤2.5的颗粒物中, 故用SOR和NOR表征PM2.5中SO42-与NO3-的转化率, NOR与SOR越高, 表明二次转化效率越高[16], 公式如下:
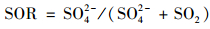
|
(2) |
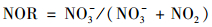
|
(3) |
依据公式 (2)、(3) 计算2015年1~12月北京大气中SOR和NOR, 按月份别对NH3和NH4+, NO2、NO和NO3-以及SO2和SO42-做图, 结合SOR、NOR、O3分析SNA与前体气体的变化规律和相互关系.2015年北京城区温湿度月均值见表 4.
|
|
表 4 2015年北京城区温湿度统计表 Table 4 Temperature and humidity statistics in the urban area of Beijing during 2015 |
温湿度增加和太阳辐射增强可促进SO2与大气中氧化剂的气相氧化反应的进行, 因此4~8月, 随着温湿度升高、太阳辐射增强、O3浓度升高, SOR不断提高, 并在8月达到峰值, 为0.82.此时SO42-最高为14.1 μg ·m-3, SO2最低为2.9 μg ·m-3, 见图 8. 9、10月随着温湿度和O3浓度的降低, SOR下降, SO42-降低, SO2升高.采暖期SO2和SO42-均较高, 尤其11、12月频发重污染, 此时O3浓度较低, SOR上升一方面考虑是大气颗粒物表面的非均相反应生成SO42-[37, 38], 另一方面考虑燃烧直接排放和污染物远距离传输的作用.

|
图 8 SO2、SO42-、O3质量浓度及SOR变化趋势 Fig. 8 Variation of SOR and concentrations of SO2, SO42- and O3 |
NH4NO3具有较强的挥发性, 温度高于30℃时, 有利于NH4NO3的分解, 温度低于15℃时, 有利于NH3与HNO3反应形成颗粒态NH4NO3[5, 39].北京5~9月, 月平均温度高于20℃, NH4NO3易分解, 此时NO3-浓度较低, NOR较低, 7月最低为0.12;其他月份温度低于15℃, 有利于NO2向NO3-转化, 且温度低时NO3-寿命相对较长, 此时NO3-浓度较高, NOR较高, 11月最高为0.26. NO2和NO3-基本呈正相关, 如图 9所示.

|
图 9 NOx、NO3-质量浓度及NOR变化趋势 Fig. 9 Variation of NOR and concentrations of NOx and NO3- |
北京大气中NH4+和 (NO3-+2SO42-) 正负离子基本平衡, 说明NH3是充足的.在大气中NH3充足的情况下, 作为受体, NH4+浓度的高低主要受酸性气体NO2、SO2影响, NO2、SO2越多, 则反应生成更多的NH4NO3和 (NH4)2SO4, 反之亦然, 如图 10.

|
图 10 NO2、SO2、NH3及NH4+质量浓度变化趋势 Fig. 10 Variation of concentrations of NO2, SO2, NH3 and NH4+ |
(1)2015年1月~12月, 对北京市城区开展为期一年的前体气体及PM2.5中SNA的研究. SO2、NOx和SNA较2014年均有所下降; NH3的年平均浓度为21.5 μg ·m-3, 高于大部分地区.按季节分析, NOx和SO2均冬季最高, 夏季最低; NH3为夏季最高, 秋冬较低; NH4+浓度和体积分数四季差异均不大, NO3-浓度和体积分数均以夏季最低, SO42-浓度为冬季最高, 体积分数为夏季占比最大.
(2) 整个观测期间, ([NO3-]+2[SO42-]) 和NH4+平衡性良好, 且两者比值小于1, 说明NH4+过量, NH3充足, 阴离子主要以NO3-和SO42-的形式存在.夏季 ([NO3-]+2[SO42-])/[NH4+]比值略大于1.0, 是NO3-除与NH4+结合, 还可与Ca2+、Mg2+和Na+等阳离子结合的原因.
(3) 从优级天到严重污染天气, 各前体气体及SNA的质量浓度均呈上升趋势, 其中NOx增加最迅猛.重度污染到严重污染, SO2有所下降. NH4+的贡献率维持在相对稳定的水平, SO42-在轻度污染级别贡献率最大, NO3-是重污染过程累积效应比较明显且贡献率最大的离子.重污染中, 颗粒物表面的非均相转化对SO42-和NO3-的形成起到更为重要的作用.
(4) 北京大气PM2.5中NO3-、SO42-和NH4+与其前体气体均显著相关.受SOR影响, SO2和SO42-的浓度变化呈负相关. NO2和NO3-基本呈正相关, 相比NH3, NH4+浓度的高低受酸性气体NO2、SO2影响更大.
| [1] | 王占山, 孙峰, 邱启鸿, 等. 冬季北京城区大气重污染特征分析[J]. 环境科技, 2015, 28(2): 47–53. Wang Z S, Sun F, Qiu Q H, et al. Analyzing characteristics of heavy air pollution events during the winter in urban beijing[J]. Environmental Science and Technology, 2015, 28(2): 47–53. |
| [2] | He K B, Yang F M, Ma Y L, et al. The characteristics of PM2^5 in Beijing, China[J]. Atmospheric Environment, 2001, 35(29): 4959–4970. DOI: 10.1016/S1352-2310(01)00301-6 |
| [3] | He K, Zhao Q, Ma Y, et al. Spatial and seasonal variability of PM2^5 acidity at two Chinese megacities:insights into the formation of secondary inorganic aerosols[J]. Atmospheric Chemistry and Physics, 2012, 12(3): 25557–25603. |
| [4] | 李丽平.青岛大气PM2.5中水溶性无机离子及其前体气体的浓度特征和气-粒平衡关系[D].青岛:中国海洋大学, 2014. |
| [5] | Hassan S K, El-Abssawy A A, Khoder M I. Characteristics of gas-phase nitric acid and ammonium-nitrate-sulfate aerosol, and their gas-phase precursors in a suburban area in Cairo, Egypt[J]. Atmospheric Pollution Research, 2013, 4(1): 117–129. DOI: 10.5094/APR.2013.012 |
| [6] | Hsu Y M, Clair T A. Measurement of fine particulate matter water-soluble inorganic species and precursor gases in the Alberta Oil Sands Region using an improved semicontinuous monitor[J]. Journal of the Air & Waste Management Association, 2015, 65(4): 423–435. |
| [7] | 李丽平, 石金辉, 李非非, 等. 青岛大气中HNO3、HNO2、NH3及PM2^5中氮组分的浓度特征和气-粒平衡关系[J]. 环境科学学报, 2014, 34(11): 2869–2877. Li L P, Shi J H, Li F F, et al. Concentrations of nitric acid, nitrous acid, ammonia gases and partner nitrogen ions in PM2^5 and gas-particle partitioning analysis in Qingdao[J]. Acta Scientiae Circumstantiae, 2014, 34(11): 2869–2877. |
| [8] | Zhuang H, Chan C K, Fang M, et al. Size distributions of particulate sulfate, nitrate, and ammonium at a coastal site in Hong Kong[J]. Atmospheric Environment, 1999, 33(6): 843–853. DOI: 10.1016/S1352-2310(98)00305-7 |
| [9] | 朱彤, 尚静, 赵德峰. 大气复合污染及灰霾形成中非均相化学过程的作用[J]. 中国科学:化学, 2011, 40(12): 1731–1740. Zhu T, Shang J, Zhao D F. The roles of heterogeneous chemical processes in the formation of an air pollution complex and gray haze[J]. Scientia Sinica Chimica, 2011, 40(12): 1731–1740. |
| [10] | Behera S N, Betha R, Balasubramanian R. Insights into chemical coupling among acidic gases, ammonia and secondary inorganic aerosols[J]. Aerosol and Air Quality Research, 2013, 13(4): 1282–1296. |
| [11] | 郑晓霞, 李令军, 赵文吉, 等. 京津冀地区大气NO2污染特征研究[J]. 生态环境学报, 2014, 23(12): 1938–1945. Zheng X X, Li L J, Zhao W J, et al. Spatial and temporal characteristics of atmospheric NO2 in the Beijing-Tianjin-Hebei region[J]. Ecology and Environmental Sciences, 2014, 23(12): 1938–1945. |
| [12] | Yao X H, Ling T Y, Fang M, et al. Comparison of thermodynamic predictions for in situ pH in PM2^5[J]. Atmospheric Environment, 2006, 40(16): 2835–2844. DOI: 10.1016/j.atmosenv.2006.01.006 |
| [13] | GB 3095-2012, 环境空气质量标准[S]. |
| [14] | 王英, 李令军, 刘阳, 等. 京津冀与长三角区域大气NO2污染特征[J]. 环境科学, 2012, 33(11): 3685–3692. Wang Y, Li L J, Liu Y, et al. Characteristics of atmospheric NO2 in the Beijing-Tianjin-Hebei region and the Yangtze river delta analyzed by satellite and ground observations[J]. Environmental Science, 2012, 33(11): 3685–3692. |
| [15] | Gupta A K, Karar K, Ayoob S, et al. Spatio-temporal characteristics of gaseous and particulate pollutants in an urban region of Kolkata, India[J]. Atmospheric Research, 2008, 87(2): 103–115. DOI: 10.1016/j.atmosres.2007.07.008 |
| [16] | Shon Z, Ghosh S, Kim K H, et al. Analysis of water-soluble ions and their precursor gases over diurnal cycle[J]. Atmospheric Research, 2013, 132-133: 309–321. DOI: 10.1016/j.atmosres.2013.06.003 |
| [17] | Chiwa M. Characteristics of atmospheric nitrogen and sulfur containing compounds in an inland suburban-forested site in northern Kyushu, western Japan[J]. Atmospheric Research, 2010, 96(4): 531–543. DOI: 10.1016/j.atmosres.2010.01.001 |
| [18] | Hu M, Wu ZJ, Slanina J, et al. Acidic gases, ammonia and water-soluble ions in PM2^5at a coastal site in the Pearl River Delta, China[J]. Atmospheric Environment, 2008, 42(25): 6310–6320. DOI: 10.1016/j.atmosenv.2008.02.015 |
| [19] | Tsai J H, Chang L P, Chiang H L. Size mass distribution of water-soluble ionic species and gas conversion to sulfate and nitrate in particulate matter in southern Taiwan[J]. Environmental Science and Pollution Research, 2013, 20(7): 4587–4602. DOI: 10.1007/s11356-012-1371-5 |
| [20] | Battye W, Aneja V P, Roelle P A. Evaluation and improvement of ammonia emissions inventories[J]. Atmospheric Environment, 2003, 37(27): 3873–3883. DOI: 10.1016/S1352-2310(03)00343-1 |
| [21] | Clarisse L, Clerbaux C, Dentener F, et al. Global ammonia distribution derived from infrared satellite observations[J]. Nature Geoscience, 2009, 2(7): 479–483. DOI: 10.1038/ngeo551 |
| [22] | Wu Z J, Hu M, Shao K S, et al. Acidic gases, NH3and secondary inorganic ions in PM10 during summertime in Beijing, China and their relation to air mass history[J]. Chemosphere, 2009, 76(8): 1028–1035. DOI: 10.1016/j.chemosphere.2009.04.066 |
| [23] | Biswas K F, Ghauri B M, Husain L. Gaseous and aerosol pollutants during fog and clear episodes in south Asian urban atmosphere[J]. Atmospheric Environment, 2008, 42(33): 7775–7785. DOI: 10.1016/j.atmosenv.2008.04.056 |
| [24] | Wang H L, Zhou Y M, Zhuang Y H, et al. Characterization of PM2^5/PM2^5-10 and source tracking in the juncture belt between urban and rural areas of Beijing[J]. Chinese Science Bulletin, 2009, 54(14): 2506–2515. DOI: 10.1007/s11434-009-0021-x |
| [25] | 杨懂艳, 刘保献, 张大伟, 等. 2012~2013年间北京市PM2^5中水溶性离子时空分布规律及相关性分析[J]. 环境科学, 2015, 36(3): 768–773. Yang D Y, Liu B X, Zhang D W, et al. Correlation, seasonal and temporal variation of water-soluble ions of PM2^5 of PM2^5in Beijing during 2012-2013[J]. Environmental Science, 2015, 36(3): 768–773. |
| [26] | Gu J X, Bai Z P, Li W F, et al. Chemical composition of PM2^5 during winter in Tianjin, China[J]. Particuology, 2011, 9(3): 215–221. DOI: 10.1016/j.partic.2011.03.001 |
| [27] | 张婷, 曹军骥, 吴枫, 等. 西安春夏季气体及PM2^5中水溶性组分的污染特征[J]. 中国科学院研究生院学报, 2007, 24(5): 641–647. Zhang T, Cao J J, Wu F, et al. Characterization of gases and water soluble ion of PM2^5 during spring and summer of 2006 in Xi'an[J]. Journal of the Graduate School of the Chinese Academy of Sciences, 2007, 24(5): 641–647. |
| [28] | Wang Y, Zhuang G S, Zhang X Y, et al. The ion chemistry, seasonal cycle, and sources of PM2^5 and TSP aerosol in Shanghai[J]. Atmospheric Environment, 2006, 40(16): 2935–2952. DOI: 10.1016/j.atmosenv.2005.12.051 |
| [29] | Cheng Y, Lee S C, Ho K F, et al. Chemically-speciated on-road PM2^5 motor vehicle emission factors in Hong Kong[J]. Science of the Total Environment, 2010, 408(7): 1621–1627. DOI: 10.1016/j.scitotenv.2009.11.061 |
| [30] | Qin Y J, Eugene K, Hopke P K. The concentrations and sources of PM2^5 in metropolitan New York City[J]. Atmospheric Environment, 2006, 40(S2): 312–332. |
| [31] | Kang C M, Lee H S, Kang B W, et al. Chemical characteristics of acidic gas pollutants and PM2^5species during hazy episodes in Seoul, South Korea[J]. Atmospheric Environment, 2004, 38(28): 4749–4760. DOI: 10.1016/j.atmosenv.2004.05.007 |
| [32] | Baek B H, Aneja V P. Measurement and analysis of the relationship between ammonia, acid gases, and fine particles in Eastern North Carolina[J]. Journal of the Air & Waste Management Association, 2004, 54(5): 623–633. |
| [33] | 廖晓农, 张小玲, 王迎春, 等. 北京地区冬、夏季持续性雾霾发生的环境气象条件比对分析[J]. 环境科学, 2014, 35(6): 2031–2044. Liao X N, Zhang X L, Wang Y C, et al. Comparative analysis on meteorological condition for persistent haze cases in summer and winter in Beijing[J]. Environmental Science, 2014, 35(6): 2031–2044. |
| [34] | Saylor R D, Edgerton E S, Hartsell B E, et al. Continuous gaseous and total ammonia measurements from the southeastern aerosol research and characterization (SEARCH) study[J]. Atmospheric Environment, 2010, 44(38): 4994–5004. DOI: 10.1016/j.atmosenv.2010.07.055 |
| [35] | Han Y J, Kim S R, Jung J H. Long-term measurements of atmospheric PM2^5 and its chemical composition in rural Korea[J]. Journal of Atmospheric Chemistry, 2011, 68(4): 281–298. DOI: 10.1007/s10874-012-9225-6 |
| [36] | Horemans B, Krata A, Buczynska A J, et al. Major ionic species in size-segregated aerosols and associated gaseous pollutants at a coastal site on the Belgian North Sea[J]. Journal of Environmental Monitoring, 2009, 11(3): 670–677. DOI: 10.1039/B815059A |
| [37] | Finlayson-Pitts B J. Reactions at surfaces in the atmosphere:integration of experiments and theory as necessary (but not necessarily sufficient) for predicting the physical chemistry of aerosols[J]. Physical Chemistry Chemical Physics, 2009, 11(36): 7760–7779. DOI: 10.1039/b906540g |
| [38] | Aikawa M, Hiraki T, Suzuki M, et al. Separate chemical characterizations of fog water, aerosol, and gas before, during, and after fog events near an industrialized area in Japan[J]. Atmospheric Environment, 2007, 41(9): 1950–1959. DOI: 10.1016/j.atmosenv.2006.10.049 |
| [39] | Galindo N, Nicolás J F, Yubero E, et al. Factors affecting levels of aerosol sulfate and nitrate on the Western Mediterranean coast[J]. Atmospheric Research, 2008, 88(3-4): 305–313. DOI: 10.1016/j.atmosres.2007.11.024 |