 2017, Vol. 38
2017, Vol. 38

2. 中国科学院大学, 北京 100049;
3. 中南林业科技大学生命科学与技术学院, 长沙 410004;
4. 中国科学院亚热带农业生态研究所公共技术服务中心, 长沙 410125
 , WEI Xiao-meng1 , WU Xiao-hong1,3 , YUAN Hong-zhao1,4
, WEI Xiao-meng1 , WU Xiao-hong1,3 , YUAN Hong-zhao1,4 , WANG Jiu-rong1,4 , LI Yu-yuan1 , GE Ti-da1 , WU Jin-shui1
, WANG Jiu-rong1,4 , LI Yu-yuan1 , GE Ti-da1 , WU Jin-shui1
2. University of Chinese Academy of Sciences, Beijing 100049, China;
3. Faculty of Life Science and Technology, Central-South University of Forestry and Technology, Changsha 410004, China;
4. The Public Service Technology Center, Institute of Subtropical Agriculture, Chinese Academy of Sciences, Changsha 410125, China
自养微生物能够利用简单的无机物作为营养物质进行正常生长代谢活动,广泛分布于各生态系统中.土壤对大气CO2的固定主要是土壤自养微生物参与的同化过程[1].稻田是我国典型的农田生态系统,我国现有稻田面积340多万hm2,约占全国耕地总面积的27%[2].IPCC报告两次指出[3, 4],包括稻田生态系统在内的农田生态系统对扼制大气CO2浓度升高发挥着不可忽视的作用.据估算,农田土壤微生物的年碳同化速率为100~450 kg ·hm-2,其对全球碳循环的贡献率为0.9%~4.1%[2, 5, 6].
迄今为止发现的5条固碳途径中,卡尔文循环是自养微生物同化CO2的最主要的途径[7].该过程中起关键作用的酶是1, 5-二磷酸核酮糖羧化酶/加氧酶(RubisCO).根据RubisCO大亚基氨基酸序列同源性、空间结构多样性、催化性能以及对O2的敏感程度,可将其分为Form Ⅰ、Form Ⅱ、Form Ⅲ和Form Ⅳ这4种类型[8].其中Form Ⅰ存在于藻类、蓝细菌、全部陆地植物和绝大多数好氧光能及化能自养微生物中,Form Ⅱ存在于数种鞭毛藻类、光合细菌和好氧及兼性厌氧化能自养细菌中,Form Ⅲ仅存在于古菌中,Form Ⅳ在氨基酸序列和三级结构上与其他RubisCO相似,但对1, 5-二磷酸核酮糖的羧化或氧化过程并无催化作用[9].cbbL和cbbM分别是RubisCO Form Ⅰ和Form Ⅱ的编码基因,由于其具有高度保守性,常作为研究不同环境中卡尔文循环自养固碳微生物群落多样性研究的标记物[10~12].目前,借助现代分子生物学技术,通过分析环境样品中功能基因的多样性进而研究自养微生物固碳的分子机制及其对不同生境的响应和反馈已成为这一领域的研究热点.
RubisCO在土壤C固定过程中发挥重要的作用.在本实验室的前期研究中,通过功能基因cbbL分子标记技术,发现稻田土壤cbbL具有很高的多样性,细菌cbbL丰度与碳同化速率呈显著正相关关系(r=0.903)[13, 14].此外,大量研究表明,碳同化微生物对土壤特性和环境因子变化比较敏感,植被类型、土壤有机质含量、土壤质地、施肥方式、根际效应、光照和深度等因素对土壤cbbL和cbbM的多样性和丰度均有显著影响[6, 14~19].然而,以往的研究很少同时对cbbL和cbbM两种基因的数量与种群结构多样性进行研究,无法将两者的数量与多样性进行同步分析和比较.
本研究选取4种典型水稻土,通过室内培养,结合DNA水平上的分子生物学研究手段(qPCR、克隆测序、T-RFLP等),对卡尔文循环功能基因cbbL和cbbM的丰度和群落结构多样性进行研究,探讨典型水稻土中的固碳微生物数量和系统发育关系,以期为水稻土的固碳潜力及稻田可持续管理提供数据支撑和理论依据.
1 材料与方法 1.1 供试土壤采集选取中国南方4个不同水稻产区4种典型稻田土壤:江西鹰潭(P1)、浙江嘉兴(P2)、湖南桃源古市(P3)和湖南桃源宝洞峪(P4)为供试土壤.将2012年7月早稻收割后采集的耕层土壤(0~20 cm)风干,用于室内培养实验.风干土壤样品分别过0.25 mm和0.149 mm筛,用于测定土壤基本理化性质.供试土壤基本理化性质见表 1.
|
|
表 1 4种供试水稻土壤的基本理化性质1) Table 1 Basic physico-chemical properties of four paddy soils used in this study |
1.2 室内培养实验
为了排除自然环境因素的影响,本研究采用室内培养方式进行.培养实验开始前,将每种土样的200 g风干土的含水量调节至45%,密封放于25℃恒温培养室2周使微生物复苏.然后将土样等量分装于50 mL离心管中,加入ddH2O充分混匀,静止沉淀30 min,调整液面使所有土壤淹水2 cm,每种土壤设置3个重复.所有离心管置于恒温培养箱中,管壁裹上锡箔纸使光照方向为正上方,25℃连续培养45 d.培养过程中每天光照12 h (光照强度4 000 lx),黑暗12 h.培养结束后的土壤样品均匀混合用液氮冷冻后保存在-80℃冰箱.
1.3 土壤理化性质测定pH测定以水为浸提剂,水土比为2.5 :1;阳离子交换量采用乙酸铵交换法测定;含水量在105℃烘箱中过夜干燥至恒重后测定;土壤SOC采用元素分析仪测定;土壤MBC、MBN采用氯仿熏蒸-0.5 mol ·L-1 K2SO4浸提法测定.
1.4 土壤DNA提取土壤总DNA的提取采用经适当修改的SDS-GITC-PEG法[13, 20].提取后的DNA用1%琼脂糖凝胶电泳和紫外检测仪(NanoDrop ND-1000, Germany)进行浓度和质量检测.
1.5 定量PCR用qPCR对不同稻田土壤细菌和古菌16S rRNA及固碳功能基因cbbL、cbbM的丰度进行分析.引物信息如下:细菌16S rRNA:Eub-8F (5′-AGAGTTTGATCCTGGCTCAG-3′),Eub-926R (5′-CCGTCAATTCCTTTRAGTTT-3′);古菌16S rRNA:Arc109F (5′-ACKGCTCA-GTAACACGT-3′),Arc934R (5′-GTGCTCCCCCGCCAATTCCT-3′);cbbL:k2f (5′-ACCAYCAAGCCSAAGCTSGG-3′),v2r (5′-GCCTTCSAGCTTGCCSACCRC-3′)[11];cbbM:cbbM-F (5′-GGCACCATCATCAAGCCCAAG-3′),cbbM-R (5′-TCTTGCCGTAGCCCA-TGGTGC-3′)[12];qPCR体系含SYBR Premix ExTaq I (TaKaRa Biotechnology, Japan)5 μL,前后引物各0.5 μL,模板DNA (稀释至5 ng·μL-1)1 μL,补无菌ddH2O水至10 μL.反应条件为:95℃ 5 min;95℃ 30 s,52℃ 45 s,72℃ 1 min,35个循环;72℃终延伸10 min.取通过测序、序列比对确定序列正确性的阳性克隆子进行扩大培养,提取质粒DNA,紫外检测仪检测浓度后以10倍梯度稀释作为标准样品,制作标准曲线.所扩增的溶解曲线都为单峰,说明引物特异性良好,扩增效率为90%~110%,标线R2均大于0.990.
1.6 末端限制性长度多态性分析(T-RFLP)采用T-RFLP技术研究不同土壤样品中卡尔文循环功能基因cbbL和cbbM的多样性差异,引物分别为k2f/v2r和cbbM-F/cbbM-R,其中正向引物5′端用6-羧基二乙酸荧光素(FAM)标记.反应体系如下:TaqDNA聚合酶Mix (全式金,中国)25 μL,正向和反向引物各1 μL,模板50~100 ng,ddH2O补水至50 μL.cbbL PCR条件为:95℃ 5 min;95℃ 30 s,66℃ 45 s,72℃ 1 min,退火温度每1个循环降1℃,5个循环;95℃ 30 s,62℃ 45 s,72℃ 1 min,30个循环;72℃延伸10 min.cbbM PCR条件为:95℃ 5 min;95℃ 30 s,57℃ 30 s,72℃ 1 min,35个循环;72℃延伸10 min.扩增的PCR产物用琼脂糖凝胶纯化试剂盒(天根,中国)纯化,纯化后的产物分别用常规型限制性内切酶Hha Ⅰ、Msp Ⅰ(TaKaRa Biotechnology,Japan)进行消化,操作步骤按照说明书进行.酶切产物送至上海桑尼生物技术有限公司进行自动测序分析(Model 373A, Applied Biosystems, Weiterstadt, Germany).相差±1 bp的片段归为同一片段,参照文献[21]的方法进行相对丰度计算,所有丰度>1%的片段为有义片段并纳入分析,>10%的片段定义为该样品的优势种群[13].生物多样性用香农指数(H)和Pielou均匀度指数(E)评价,H指数计算公式为:
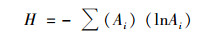
|
式中,Ai为一种片段长度的相对丰度.以H指数的计算结果为基础,E=H/Hmax,其中Hmax=lnS,S为不同片段的种类数[13, 22].
1.7 克隆、测序和系统发育分析将培养45 d后的土样DNA的3个重复混合均匀作为模板,进行PCR扩增,引物及反应条件同1.5节,PCR体系同1.6节.扩增产物用凝胶回收试剂盒(天根,中国)纯化后连入pGEM-T Easy载体,再导入感受态细胞Escherichia coli DH5α(天根,中国),经载体特异引物T7/SP6扩增检验后,每种水稻土cbbL基因随机挑取100个阳性克隆子,cbbM基因挑取50个阳性克隆进行测序.测序反应由北京六合华大基因科技有限公司广州分公司完成.利用mothur软件将相似性大于97%的序列归为同一操作分类单元(OTU),用BLAST和NCBI数据库进行序列比对,用MEGA 6.0构建系统发育树.同时分别计算各土样克隆文库的覆盖率(coverage,C),计算公式为:
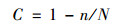
|
式中,n代表每种土样中仅有1个克隆子的OTU数,N代表每种土样克隆总数.选择有两条及以上序列的主要OTU,采用邻接法(neighbor-joining)构建系统发育树(1 000次重复计算bootstrap值).所获得的cbbL和cbbM基因序列提交至GenBank中,同一OTU提交一条代表性序列,序列登录号KX265792-KX265981(cbbL),KX265735-KX265791(cbbM).
1.8 数据分析数据处理采用Excel 2007和SPSS 19.0,不同土壤之间的差异显著性分析用单因素的邓肯法,培养时间(第0 d、第45 d)的差异显著性采用t检验.群落多样性和基因与环境因子的关系采用CANOCO 4.5软件分析,基于DCA的结果,选用RDA进行关联分析,采用手动前置选择和蒙特卡洛置换检验进行显著性分析(P < 0.05).
2 结果与分析 2.1 不同水稻土碳同化功能基因的绝对丰度和相对丰度以基因拷贝数作为碳同化功能基因的绝对丰度,结果如图 1(a)~1(d)所示.培养前(D0),土壤细菌16S rRNA丰度范围为1.01×1010~1.40×1010 copies ·g-1,其中P1最低,P4最高;古菌拷贝数P1最低,最高出现在P3.4种水稻土的cbbL丰度差异较大,其中P2显著较高,达1.78×108 copies ·g-1,其次为P1、P3,P4最低,仅0.74×108 copies ·g-1.cbbM在P3中的丰度显著较高,在其他3种土中无显著差异.培养45 d (D45)后,细菌和古菌16S rRNA拷贝数在P1、P2中无显著变化,在P3、P4中则出现较大幅度的提升,培养结束时,其丰度顺序依次为P3>P4>P2>P1.与培养前相比,4种土中cbbL和cbbM的丰度均大幅升高,cbbL为2.48×108~3.44×108 copies ·g-1,比cbbM高3个数量级,其中P1中两种功能基因丰度均为最低,cbbL在P4中最高,cbbM在P2中最高.除了P1和P2的细菌 16S rRNA 以及P1和P3的cbbM拷贝数,培养45 d后微生物数量都是显著增加的.

|
(a)细菌,(b)古菌,(c)cbbL,(d)cbbM,(e)cbbL与细菌丰度的比值,(f)cbbM与细菌丰度的比值;不同大写字母表示培养前(D0)与培养后(D45)土壤之间差异显著,不同小写字母表示不同供试土壤之间差异显著(P < 0.05) 图 1 4种不同水稻土中基因丰度以及cbbL、cbbM与细菌16S rRNA拷贝数的比值 Fig. 1 Gene abundances of four different paddy soils and the ratio of cbbL, cbbMcopies to the bacterial 16S rRNA gene |
培养前和培养后碳同化功能基因的相对丰度以功能基因拷贝数占细菌16S rRNA的百分比表示[图 1(e)~1(f)].培养前cbbL相对丰度在P1、P2中最高,分别为1.47%和1.37%.其次为P3,在P4中最低,仅0.53%.经过45 d的培养,4种土中cbbL的相对丰度均显著增加,其大小依次为P2>P1>P4>P3,其中初始相对丰度最高的P2增加幅度最大,达培养前的1.51倍,P3的增加幅度最小.cbbM初始土样的相对丰度依次为P3>P1>P4>P2.培养过程cbbM相对丰度极大地增加,且促进作用的强度随初始相对丰度的增大而减小.培养结束时,P2的相对丰度高达培养前的21.85倍,P3则仅为培养前的66%.
2.2 多样性和系统发育分析cbbL的Hha Ⅰ酶切T-RFLP图谱显示,从4种水稻土中共获得25种不同长度的末端片段,其中58bp (丰度9.3%~20.4%)和125bp (丰度13.9%~34.8%)的片段所代表的物种为所有土样共有的优势种群.58 bp片段在P2中的丰度略低于10%,在另外3种土壤中均超过15%,最高值出现在P1;125bp片段在P4中丰度最高,在P1~P3中均低于20%.此外,还存在部分土样共有或单一土样特有的优势种群,其中25bp为P1、P3共有,44bp为P1、P2共有,118bp和165bp为P3特有,133bp为P4特有[图 2(a)].

|
(a)cbbL; (b)cbbM 图 2 4种不同水稻土壤中cbbL、cbbM的限制性片段的相对丰度 Fig. 2 Average relative abundances of marker genes T-RFs in four different paddy soils for cbbL, cbbM |
4种水稻土cbbM基因的Msp Ⅰ酶切T-RFLP共获得14种不同的末端片段,其中共有的优势种群是179bp,所占比例为48.6%~90.6%,丰度高低依次为P3>P4>P1>P2,320bp (40.4%)、177bp (47.2%)、31bp (15.7%)片段所代表的物种分别为P1、P2和P4各自特有的优势种群[图 2(b)].
从4种水稻土的cbbL克隆文库中共获得190个OTU,克隆文库覆盖率分别为63%(P1)、47%(P2)、52%(P3)和85%(P4),其中40%左右的OTU形成了4个未知Cluster簇(Ⅰ~Ⅳ),余下的序列与α-Proteobacteria (17%)、β-Proteobacteria (11%)和Actinobacteria (11%)的微生物具有较高相似度,还有21%的序列与α-Proteobacteria、β-Proteobacteria的微生物都具有较高相似度.P2获得的阳性克隆子明显聚集在ClusterⅡ和mixed-Proteobacteria处,P1、P3主要分布于Actinobacteria,属于ClusterⅠ和α-Proteobacteria的物种主要存在于P1、P3和P4(图 3).T-RFLP图谱结合系统发育分析的结果可以发现,165/166bp片段的微生物主要分布在ClusterⅠ.

|
括号中P1~P4代表 4种典型水稻土,其前后的数字分别代表克隆子的数目和Hha Ⅰ模拟酶切片段长度,比例尺代表进化距离5%的序列变异 图 3 基于部分cbbL序列的系统发育树 Fig. 3 Phylogenetic tree based on partial cbbL sequences |
从所有水稻土的cbbM序列中共获得57个OTU,克隆文库覆盖率分别为81%(P1)、84%(P2)、80%(P3)和80%(P4),其中67%的序列形成2个未知Cluster簇,余下的序列与α-Proteobacteria (17%)、β-Proteobacteria (8%)和γ-Proteobacteria (8%)相似度较高.P1中的cbbM相关物种主要属于α-Proteobacteria Ⅰ,P2、P3和P4则主要属于Cluster Ⅰ、Cluster Ⅱ和α-Proteobacteria (图 4).模拟酶切结果显示,182bp片段的微生物主要分布在Cluster Ⅰ、Cluster Ⅱ和γ-Proteobacteria.

|
括号中P1~P4代表 4种典型水稻土,其前后的数字分别代表克隆子数目和Msp Ⅰ模拟酶切片段长度,比例尺代表进化距离5%的序列变异 图 4 基于部分cbbM序列的系统发育树 Fig. 4 Phylogenetic tree based on partial cbbM sequences |
根据T-RFLP图谱中末端限制性片断的种类及其峰高值,计算了4种典型稻田土壤中cbbL和cbbM基因的多样性指数和均匀度指数.从表 2中可以看出,所有样品中,cbbL的多样性指数均显著高于cbbM.其中P3中香农指数和均匀度最高;P1和P2中cbbM多样性指数均显著高于P3和P4.
|
|
表 2 基于T-RFLP数据的4种典型水稻土样的多样性指数1) Table 2 Diversity index of four typical paddy soils based on T-RFLP data |
2.4 RDA分析
对4种水稻土cbbL和cbbM的T-RFLP结果进行RDA分析,结果显示,前两轴分别可解释cbbL和cbbM多样性变化的67.5%和68.7%.粉粒含量(P=0.006)、CEC (P=0.002)、pH (P=0.002)和SOC (P=0.012)与含cbbL基因的微生物群落组成显著相关[图 5(a)];cbbM基因的微生物群落组成则受到砂粒含量(P=0.004)、SOC (P=0.002)、CEC (P=0.004)和黏粒含量(P=0.028)的显著影响[图 5(b)].

|
图 5 卡尔文循环功能基因cbbL、cbbM的T-RFLP数据与环境因子RDA分析 Fig. 5 Redundancy analysis of T-RFLP of the marker genes of Calvin cycle for cbbL T-RFLP profiles, cbbM T-RFLP profiles |
通过45 d的室内培养,4种水稻土中均检测出高丰度的cbbL基因以及较低丰度的cbbM基因,说明稻田土壤中的确存在相当数量的自养微生物.有研究表明,不同的土地利用方式和土壤类型会影响微生物数量及其群落多样性[23, 24].多样性指数是评价不同土壤微生物群落多样性的有效手段,高的香农指数和均匀度表明高的微生物群落多样性.cbbL在P3的丰度较低,但香农指数和均匀度均最高,说明微生物群落多样性与丰度没有明显的正相关关系.培养45 d后,两种功能基因的相对丰度均有增加,其中cbbL在有机质含量较低的P1和P2土壤中最高,而cbbM则在pH较高的P2和P4中较高.Yuan等[17]的最新研究发现cbbL/16S rRNA的比值在旱地土壤中最高,认为贫瘠土壤有利于碳同化微生物中的活性表达;而cbbM基因丰度表现为P2>P4>P3>P1,pH大的土壤中该基因丰度较高,其与细菌16S rRNA的比值可能与cbbM类群微生物受pH影响有关.
4种稻田土壤都含有共有的及各自特有的cbbL和cbbM优势种群(图 2).碳同化自养微生物有光能自养微生物和化能自养微生物两种,克隆文库分析结果显示cbbL的阳性克隆子多为化能自养菌,主要与变形菌的慢生根瘤菌、维氏硝酸杆菌、亚硝化螺菌和硫杆菌等的序列相似,也有一部分与固氮红细菌等一些不产氧光合细菌聚类;cbbM的阳性克隆子与变形菌门的硫化菌等专性化能自养菌的相似度较高.Wu等[6]的研究显示光照主要影响表层1 cm左右的碳同化自养微生物,化能自养菌以CO2为主要碳源,通过氧化无机物获得能量进行自养生长,成为了在光照条件不足的土层中的优势物种.Yuan等[5]的研究发现,稻田土壤碳同化细菌主要是不产氧兼性自养菌,如变形菌门的红假单胞菌、慢生根瘤菌和固氮红螺菌等,与本文结果较为一致.同时,P2中的cbbL和P1中的cbbM与其他样品中的阳性克隆子系统发育关系较远,说明不同的土壤中的碳同化自养微生物物种差异较大(图 3~4),cbbL克隆文库显示还存在有放线菌门的分枝杆菌等一些新的自养固碳微生物类群,还有待进一步深入研究.
RDA分析结果显示4种不同稻田土壤分别聚集在二维排序图的不同位置,说明不同稻田土壤cbbL和cbbM群落结构存在差异,SOC和CEC是其显著影响因素(图 5),这与Yuan等[14]和Xiao等[24]的研究结果一致,CEC可以直接影响土壤肥力及其对微生物元素的可利用性[25];Yuan等对不同施肥条件下固碳细菌的研究发现SOC也对固碳细菌丰度和群落结构有显著影响,这可能是由于其降解能为微生物提供各类无机元素等营养物质和能量.此外,pH可以通过H+浓度改变土壤中营养元素的形态从而影响自养微生物类群[26].另外,黏粒含量也是影响固碳微生物多样性和丰度变异的重要因素,这可能与一般更小的土壤组分中常常有利于有机碳积累和基因多样性的存在的原因有关[27].Selesi等[28]发现cbbL基因主要分布在黏粒和粉粒中,而在粗颗粒中几乎检测不到cbbL基因,P1黏粒含量最低,砂粒含量最高,土壤粗颗粒组分中营养物质难以积累,不利于自养微生物的生长,因此16S rRNA以及卡尔文功能基因拷贝数量最低,而P3、P4土壤中黏粒含量较高,因而微生物数量相对较高.不同的施肥措施和土地利用方式使得土壤环境和养分发生改变从而影响了对环境变化敏感的自养微生物的生长和代谢活动,最终导致碳同化功能微生物种群结构的变化.本文研究表明,土壤性质的不同会导致固碳功能基因微生物多样性和丰度的变化,基于此,可以采取一定的田间管理措施来增强自养固碳微生物的活性和多样性.
4 结论(1)稻田土壤室内培养实验表明,在没有其他外来扰动的条件下,碳同化微生物数量会随着时间增加,卡尔文循环关键酶RubisCO的2种编码基因(cbbL和cbbM)丰度范围(干土中)为105~108 copies ·g-1,cbbL丰度比cbbM高3个数量级,且多样性高于cbbM.
(2)不同稻田土壤的碳同化微生物群落组成存在明显差异,且具有共有和各自特有的优势种群,这些微生物多为变形菌和放线菌.
(3)土壤性质的不同会导致固碳微生物功能基因丰度和多样性的变化,SOC和CEC是编码cbbL和cbbM基因的功能微生物群落结构的显著影响因子.
| [1] | 陈晓娟, 吴小红, 简燕, 等. 农田土壤自养微生物碳同化潜力及其功能基因数量、关键酶活性分析[J]. 环境科学, 2014, 35(3) : 1144–1150. Chen X J, Wu X H, Jian Y, et al. Carbon dioxide assimilation potential, functional gene amount and RubisCO activity of autotrophic microorganisms in agricultural soils[J]. Environmental Science, 2014, 35(3) : 1144–1150. |
| [2] | Ge T D, Yuan H Z, Zhu H H, et al. Biological carbon assimilation and dynamics in a flooded rice-soil system[J]. Soil Biology and Biochemistry, 2012, 48 : 39–46. DOI: 10.1016/j.soilbio.2012.01.009 |
| [3] | IPCC. Climate Change 1995:The Science of Climate Change[M]. Cambridge, UK: Cambridge University Press, 1995. |
| [4] | IPCC. Climate Change 2007:Impacts, Adaptation and Vulnerability. Contribution of Working Group Ⅱ to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change[M]. Geneva, Switzerland: Cambridge University Press, 2007. |
| [5] | Yuan H Z, Ge T D, Chen C Y, et al. Significant role for microbial autotrophy in the sequestration of soil carbon[J]. Applied and Environmental Microbiology, 2012, 78(7) : 2328–2336. DOI: 10.1128/AEM.06881-11 |
| [6] | Wu X H, Ge T D, Yuan H Z, et al. Changes in bacterial CO2 fixation with depth in agricultural soils[J]. Applied Microbiology and Biotechnology, 2014, 98(5) : 2309–2319. DOI: 10.1007/s00253-013-5179-0 |
| [7] | Berg I A. Ecological aspects of the distribution of different autotrophic CO2 fixation pathways[J]. Applied and Environmental Microbiology, 2011, 77(6) : 1925–1936. DOI: 10.1128/AEM.02473-10 |
| [8] | Tabita F R. Microbial ribulose 1, 5-bisphosphate carboxylase/oxygenase:a different perspective[J]. Photosynthesis Research, 1999, 60(1) : 1–28. DOI: 10.1023/A:1006211417981 |
| [9] | Tabita F R, Satagopan S, Hanson T E, et al. Distinct form Ⅰ, Ⅱ, Ⅲ, and Ⅳ Rubisco proteins from the three kingdoms of life provide clues about Rubisco evolution and structure/function relationships[J]. Journal of Experimental Botany, 2008, 59(7) : 1515–1524. |
| [10] | Bassham J A, Benson A A, Kay L D, et al. The path of carbon in photosynthesis. Ⅹ Ⅺ. the cyclic regeneration of carbon dioxide acceptor[J]. Journal of the American Chemical Society, 1954, 76(7) : 1760–1770. DOI: 10.1021/ja01636a012 |
| [11] | Nanba K, King G M, Dunfield K. Analysis of facultative lithotroph distribution and diversity on volcanic deposits by use of the large subunit of ribulose 1, 5-bisphosphate carboxylase/oxygenase[J]. Applied and Environmental Microbiology, 2004, 70(4) : 2245–2253. DOI: 10.1128/AEM.70.4.2245-2253.2004 |
| [12] | Alfreider A, Vogt C, Hoffmann D, et al. Diversity of ribulose-1, 5-bisphosphate carboxylase/oxygenase large-subunit genes from groundwater and aquifer microorganisms[J]. Microbial Ecology, 2003, 45(4) : 317–328. DOI: 10.1007/s00248-003-2004-9 |
| [13] | Yuan H Z, Ge T D, Zou S Y, et al. Effect of land use on the abundance and diversity of autotrophic bacteria as measured by ribulose-1, 5-biphosphate carboxylase/oxygenase (RubisCO) large subunit gene abundance in soils[J]. Biology and Fertility of Soils, 2013, 49(5) : 609–616. DOI: 10.1007/s00374-012-0750-x |
| [14] | Yuan H Z, Ge T D, Wu X H, et al. Long-term field fertilization alters the diversity of autotrophic bacteria based on the ribulose-1, 5-biphosphate carboxylase/oxygenase (RubisCO) large-subunit genes in paddy soil[J]. Applied Microbiology and Biotechnology, 2012, 95(4) : 1061–1071. DOI: 10.1007/s00253-011-3760-y |
| [15] | Selesi D, Schmid M, Hartmann A. Diversity of green-like and red-like ribulose-1, 5-bisphosphate carboxylase/oxygenase large-subunit genes (cbbL) in differently managed agricultural soils[J]. Applied and Environmental Microbiology, 2005, 71(1) : 175–184. DOI: 10.1128/AEM.71.1.175-184.2005 |
| [16] | Yousuf B, Keshri J, Mishra A, et al. Application of targeted metagenomics to explore abundance and diversity of CO2-fixing bacterial community using cbbL gene from the rhizosphere of Arachis hypogaea[J]. Gene, 2012, 506(1) : 18–24. DOI: 10.1016/j.gene.2012.06.083 |
| [17] | Yuan H Z, Ge T D, Chen X B, et al. Abundance and diversity of CO2-assimilating bacteria and algae within red agricultural soils are modulated by changing management practice[J]. Microbial Ecology, 2015, 70(4) : 971–980. DOI: 10.1007/s00248-015-0621-8 |
| [18] | Miltner A, Kopinke F D, Kindler R, et al. Non-phototrophic CO2 fixation by soil microorganisms[J]. Plant and Soil, 2005, 269(1) : 193–203. |
| [19] | Xiao K Q, Nie S A, Bao P, et al. Rhizosphere effect has no effect on marker genes related to autotrophic CO2 fixation in paddy soils?[J]. Journal of Soils and Sediments, 2014, 14(6) : 1082–1087. DOI: 10.1007/s11368-014-0864-x |
| [20] | Chen Z, Luo X Q, Hu R G, et al. Impact of long-term fertilization on the composition of denitrifier communities based on nitrite reductase analyses in a paddy soil[J]. Microbial Ecology, 2010, 60(4) : 850–861. DOI: 10.1007/s00248-010-9700-z |
| [21] | Lukow T, Dunfield P F, Liesack W. Use of the T-RFLP technique to assess spatial and temporal changes in the bacterial community structure within an agricultural soil planted with transgenic and non-transgenic potato plants[J]. FEMS Microbiology Ecology, 2000, 32(3) : 241–247. DOI: 10.1111/fem.2000.32.issue-3 |
| [22] | Shannon C E. A mathematical theory of communication[J]. Acm Sigmobile Mobile Computing and Communications Review, 2001, 5(1) : 3–55. DOI: 10.1145/584091 |
| [23] | 沈冰洁, 祝贞科, 袁红朝, 等. 不同种植方式对亚热带红壤微生物多样性的影响[J]. 环境科学, 2015, 36(10) : 3839–3844. Shen B J, Zhu Z K, Yuan H Z, et al. Effects of different plantation type on the abundance and diversity of soil microbes in subtropical red soils[J]. Environmental Science, 2015, 36(10) : 3839–3844. |
| [24] | Xiao K Q, Bao P, Bao Q L, et al. Quantitative analyses of ribulose-1, 5-bisphosphate carboxylase/oxygenase (RubisCO) large-subunit genes (cbbL) in typical paddy soils[J]. FEMS Microbiology Ecology, 2014, 87(1) : 89–101. DOI: 10.1111/1574-6941.12193 |
| [25] | Zhang L M, Hu H W, Shen J P, et al. Ammonia-oxidizing archaea have more important role than ammonia-oxidizing bacteria in ammonia oxidation of strongly acidic soils[J]. The ISME Journal, 2012, 6(5) : 1032–1045. DOI: 10.1038/ismej.2011.168 |
| [26] | Stockdale E A, Shepherd M A, Fortune S, et al. Soil fertility in organic farming systems-fundamentally different?[J]. Soil Use and Management, 2002, 18(1) : 301–308. |
| [27] | Balesdent J, Chenu C, Balabane M. Relationship of soil organic matter dynamics to physical protection and tillage[J]. Soil and Tillage Research, 2000, 53(3-4) : 215–230. DOI: 10.1016/S0167-1987(99)00107-5 |
| [28] | Selesi D, Pattis I, Schmid M, et al. Quantification of bacterial RubisCO genes in soils by cbbL targeted real-time PCR[J]. Journal of Microbiological Methods, 2007, 69(3) : 497–503. DOI: 10.1016/j.mimet.2007.03.002 |