 2017, Vol. 38
2017, Vol. 38


2. 中国科学院地球环境研究所, 黄土与第四纪地质国家重点实验室, 西安 710061
2. State Key Laboratory of Loess and Quaternary Geology, Institute of Earth Environment, Chinese Academy of Sciences, Xi'an 710061, China
二次有机气溶胶(SOA)是对流层大气中一类重要污染物,在2.5 μm以下的细颗粒物上,占颗粒物总质量高达75%[1],是构成大气霾的主要物质之一[2, 3].二元羧酸类SOA是大气中普遍存在的一类重要水溶性SOA (WSOA)[4],其蒸气压较低,很容易富集在气溶胶颗粒物上,且二元羧酸水溶性较强,极易溶于水,从而影响气溶胶颗粒物的吸湿性,促进水汽在颗粒物表面凝结,进而影响云凝结核(CCN)的活性及颗粒物的粒径分布,改变云的光学性质与寿命,使得低云、云量增多,从而使云的反射率增加,减少入射辐射[5, 6].
近20年来,国内外学者对二元羧酸类SOA的来源和形成机制开展了大量研究,并取得了一定的进展. 1996年,Kawamura等[7]最早提出了大气中乙二醛(Gly)可以氧化生成草酸(C2,含量最高的二元羧酸),但并未提出该反应过程的形成机制.后来的学者通过分析海洋大气云雾中的二元羧酸初步探讨了C2的液相形成机制,但并未区分其前体物的来源(人为源或生物源)的类型[8, 9].有研究表明,mGly和Gly (两个最小的α-二羰基化合物)作为C2的重要前体物,主要是由生物源排放的VOCs (异戊二烯、单萜烯等)和人为源排放的VOCs (芳香烃、丙酮和乙炔等)氧化形成的[10].模式模拟结果显示,全球范围内mGly和Gly的排放量分别为140 Tg·a-1和45 Tg·a-1,其中79%的mGly和47%的Gly来自生物源异戊二烯的氧化[11].
目前,有关大气中二元羧酸类化合物的研究主要集中在城市[12, 13]、森林[14]、海洋[15]等近地面地区,而关于自由对流层大气的研究较少,尤其关于高山森林背景地区(自由对流层)二元羧酸类SOA的的化学组成及来源的研究更少.有关自由对流层气溶胶的研究目前国际上大多通过飞机航测来开展研究,少数采用飞艇或气球来进行[9].与近地面大气相比,高山地区具有海拔高、太阳辐射强的特征,高山地区的气温与相对湿度昼夜变化较大,气溶胶更容易进入云层并形成CCN[16, 17].因此,高山地区自身独特的环境条件决定了二元羧酸类SOA的化学组成、来源、形成机制与近地面相比存在着显著的差异.再者,较飞机和气球观测相比,在高山地区使用滤膜采集大气气溶胶样品更加便捷、廉价,且具有观测周期长的优点.泰山是华北平原的最高峰,以往关于泰山气溶胶化学组分与污染程度的研究,主要侧重于春季沙尘期与夏季小麦秸秆燃烧期等典型污染事件[17~20],而关于夏季生物源对SOA的研究较少[21].泰山地区森林覆盖率高达80%,生物源二次有机气溶胶(BSOA)占OC的相对含量高达23%,生物源对SOA的贡献十分显著[21].因此,本研究于2014年夏季在泰山进行采样,并分析PM2.5样品中二元羧酸类化合物的分子组成、浓度水平,阐明高山森林地区二元羧酸类SOA的来源、形成机制,不但对认识我国霾天气的形成机制和科学治理具有重要意义,还可为全球SOA模式研究提供重要的基础数据资料.
1 材料与方法 1.1 样品采集及质量浓度分析采样地点设在泰山的日观峰气象站(海拔1 534 m;117.10°E,36.25°N),采样点周边视野开阔且无明显的污染源,能客观反映华北地区高山背景区域的大气质量状况.于2014年7~8月使用大流量PM2.5采样器(Anderson,美国)采集PM2.5样品(44个),流量为1.3 L·min-1,每个样品采集时间约为24 h (当日08:00~次日08:00).所用石英滤膜使用前均在450℃烘烤6 h,以去除有机污染物,采样期间共收集了环境空白样品共2个. PM2.5质量浓度采用重量分析法,使用电子微量天平(Mettle M3, Switzerland, 灵敏度1 μg)对采样前后的滤膜进行称重.所有滤膜在称重前都必须在恒温恒湿箱[T:(20~23)℃,RH: 35%~45%]中平衡24 h以上,确保滤膜的干燥.
1.2 二元羧酸类有机化合物分析取1/4滤膜剪碎,用10 mL超纯水超声萃取3次,每次15 min,加入0.3 mL的BF3/正丁醇溶液于100℃反应1 h.冷却后,依次加入5 mL正己烷和0.3 mL乙腈,用超纯水洗涤3次,合并有机相、氮吹浓缩至100 μL,待上机检测[7].
衍生化后的样品使用气相色谱-质谱联用仪(GC/MS)、气相色谱分析仪(GC-FID)分别进行定性、定量分析,采用DB-5MS,30 m×1.25 mm×0.25 m GC色谱柱.升温程序:起始温度50℃,保持2 min,以15℃·min-1升温至120℃,然后以5℃·min-1升至300℃,保持10 min.根据样品化合物的峰面积,采用内标法定量计算环境浓度[12].
1.3 WSOC及水溶性无机离子化学分析取1/4滤膜剪碎,加入25 mL去离子水溶解、超声萃取4次,每次15 min,随后经脱色摇床振荡1 h后静置.萃取后的水溶液用0.45 μm的水系过滤器过滤,过滤后的溶液用于WSOC和无机离子分析.取其中的5 mL过滤液采用总有机碳分析仪(Shimadzu TOC-L CPH型)进行检测.剩余约17 mL的过滤液采用Dionex-600型和Dionex-500型离子色谱仪分别分析6种阴离子(F-、Cl-、Br-、NO2-、NO3-、SO42-)与5种阳离子(Na+、NH4+、K+、Mg2+、Ca2+).
2 结果与讨论 2.1 PM2.5质量浓度、WSOC及无机离子的浓度水平与污染特征图 1为泰山夏季PM2.5质量浓度与WSOC浓度的时间序列,PM2.5质量浓度范围为4.6~74 μg·m-3,平均值为(29±16) μg·m-3,比中国城市地区的平均浓度低75%[22]. WSOC的平均浓度为(1.1±0.4) μg·m-3,仅为2006年泰山小麦秸秆燃烧期间大气PM10中WSOC浓度低11%[17], 表明生物质燃烧是WSOC的一个重要来源.所有离子浓度总和占PM2.5质量浓度的27%±7.8%,与大陆背景点(青海湖)[23]相似.无机离子的浓度及其分子组成如图 2所示,从中可见,浓度最高的是SO42-,占离子总质量浓度的56%±12%,其次是NO3-(16%±8.0%)、NH4+(9.4%±2.8%),与2009年春季泰山大气PM10中无机离子的分子组成不同(NO3->SO42->NH4+)[19].泰山地区SO42-[(4.6±3.6)μg·m-3)]、NO3-[(1.0±0.6) μg·m-3]及NH4+[(0.7±0.5) μg·m-3]的浓度与国内一些高山地区(如贡嘎山、鼎湖山及长白山)的浓度相当[24],但远低于2009年春季泰山PM10中无机离子离子的浓度(SO42-、NO3-及NH4+的浓度分别为16、20及12 μg·m-3),可能由于采样季节不同与颗粒物粒径不同导致的[19].

|
图 1 泰山夏季PM2.5气溶胶中PM2.5质量浓度与WSOC浓度的日均值时间变化序列 Fig. 1 Temporal variations of daily mean concentrations of PM2.5 mass and WSOC from Mt. Taishan in summer |

|
图 2 泰山夏季PM2.5气溶胶中无机离子的组成及总离子浓度占PM2.5质量浓度的相对含量随时间的变化趋势 Fig. 2 Temporal variations of chemical composition of the inorganic ions and the ratio of total ions and PM2.5 mass at Mt. Taishan in summer |
n(NH4+)/n(2×SO42+NO3-)比值为0.4,可推断气溶胶中没有足够的NH3中和所有的H2SO4和HNO3.另外,本研究所采集PM2.5样品,主要存在粗粒子中的Ca2+和Mg2+占离子总质量浓度的相对含量很低,分别为4.4%±2.5%、1.2%±0.6%,因此不考虑Ca2+和Mg2+对SO42-、NO3-的中和作用.由此可推断泰山地区气溶胶呈酸性,酸性条件有利于SOA的生成[25].硫酸盐能够通过相似的反应过程(如液相反应)促进SOA的生成[9],因此,SO42-与WSOC的相关性较好[R2=0.78,图 3(a)].

|
图 3 线性回归分析 Fig. 3 Linear regression analysis |
本研究分析了泰山夏季PM2.5中的二元羧酸类SOA[二元羧酸(C2~C11)、酮羧酸及α-二羰基化合物],其浓度如表 1所示,其分子组成见图 4.
|
|
表 1 泰山2014年夏季PM2.5样品中二元羧酸类SOA、BSOA的示踪物及WSOC的浓度/ng·m-3 Table 1 Concentrations of dicarboxylic acids and related SOA, BSOA tracers and WSOC in PM2.5 aerosols collected at Mt. Taishan during the summer of 2014/ng·m-3 |

|
A:二元羧酸; B:酮羧酸; C: α-二羰基化合物 图 4 泰山夏季PM2.5气溶胶中二元羧酸、酮羧酸及二羰基化合物的分子构成 Fig. 4 Molecular composition of dicarboxylic acids, ketocarboxylic acids and α-dicarbonyls in PM2.5 aerosols collected at Mt. Taishan in summer |
泰山PM2.5中二元羧酸的总浓度范围为(116~916) ng·m-3,平均值为(376±189) ng·m-3,其值低于其他高山地区(如华山、喜马拉雅山)[16, 26](表 2).另外,泰山地区二元羧酸的总浓度低于城市地区,例如北京[27]、西安[12]、印度的金奈[28]、日本的札幌[29](表 2).但是此值却高于海洋地区(如中央太平洋、南大洋)[4, 30](表 2).由此可得出,在全球范围内,泰山夏季PM2.5中二元羧酸的总浓度高于海洋地区,但低于城市和其他高山地区.与上述地区一样,二元羧酸类SOA中草酸(C2)最多(图 4),范围为47~557 ng·m-3,平均浓度为(231±135) ng·m-3. C2是分子量最小的二元羧酸,主要来自长链的二元羧酸及有机前体物(芳香烃碳氢化合物、烯烃等)的光化学氧化[5, 12],也可由化石燃料及生物质燃烧等一次源直接排放[7, 13]. C2占总二元羧酸的相对含量为62%±7.7%,其次为丙二酸(C3: 12%±4.5%)、丁二酸(C4: 5.8%±1.8%)、壬二酸(C9: 5.1%±2.5%)和邻苯二甲酸(Ph: 2.9%±1.4%).如表 2所示,这种分子组成与偏远地区是相似的,例如华山[16]、青海湖[23]和喜马拉雅山[26].但是与城市地区不同,因为在城市地区来自人为污染源的前体物较多(多环芳烃和增塑剂)[13, 31],导致邻苯二甲酸(Ph)或对苯二甲酸(tPh)的浓度远高于壬二酸,更甚者远高于C2,例如在蒙古的乌兰巴托市[31],冬季PM2.5中二元羧酸有机气溶胶分子组成的顺序为tPh>C2>C4>Ph,这是因为冬季使用褐煤和木材的炉灶进行取暖和烹饪,并且一些贫民使用含有聚酯纤维的垃圾、塑料瓶作为生活燃料,导致该区的tPh急剧上升. C9主要来自植物排放的不饱和脂肪酸经光化学氧化形成的[7].泰山的C9含量较高,主要归因于植物排放大量的不饱和脂肪酸,随后经光化学氧化反应形成的.
|
|
表 2 泰山地区二元羧酸的浓度与世界其它地区的对比1)/ng·m-3 Table 2 Comparison of dicarboxylic acid concentrations in Mt. Taishan with other sites in the world/ng·m-3 |
泰山PM2.5中酮羧酸和α-二羰基化合物的平均浓度各为(14±12) ng·m-3、(8.0±5.4) ng·m-3.有研究表明,作为二元羧酸的前体物,二者是由有机前体物经大气光化学氧化反应形成或直接由化石燃料和生物质燃烧直接排放的,最后经氧化反应生成二元羧酸[26, 30].如图 4所示,浓度最高的酮羧酸为乙醛酸(ωC2),占酮羧酸总浓度的74%±17%,其次为丙酮酸(Pyr:26%±17%). mGly和Gly的浓度各为(6.6±4.7) ng·m-3、(1.4±1.0) ng·m-3.因为mGly的来源比Gly更多,并且在气溶胶中mGly与羟基自由基(·OH)的氧化速度比Gly慢[12],导致mGly的浓度高于Gly.
2.3 二元羧酸类SOA的来源与形成机制有研究表明,C2是由各种前体物(长链的二元羧酸、乙醛酸、丙酮酸)与羟基自由基(·OH)经光化学氧化形成的[9, 11].顺丁烯二酸(M)经水合作用及C4经羟基化反应可生成羟基丁二酸(hC4),又可进一步形成C2和C3[14].由于C3在α-C上存在能被相邻的两个羧基激活的活性氢原子,所以C3在高温条件下不稳定,可经羟基丙二酸或氧代丙二酸(kC3)等中间体最后氧化生成C2[23].因此,C2/C4和C3/C4这两个比值可用来表征气溶胶的老化程度,即可用来评估一次源与二次源的重要性.泰山PM2.5中C2/C4和C3/C4比值分别为12±6.8、2.3±1.2,高于城市(中国14大城市[13]、札幌[29])及海洋地区(济州岛[15]、南大洋[30]),表明泰山地区的气溶胶氧化反应较强致使气溶胶老化程度较深.
Pavuluri等[28]认为当二元羧酸类SOA主要来自本地源时,C3/C4比值与环境温度呈较强的相关性,反之,远源输入起的作用更大.为了评估当地源与长距离传输对泰山地区气溶胶的影响,对C3/C4与环境温度进行相关性分析[图 3(b)].由图 3(b)可知C3/C4与温度间相关性较强(R2=0.67),表明泰山PM2.5中二元羧酸类SOA主要受本地源的影响.
有研究证明M主要来自芳香烃碳氢化合物(例如苯和甲苯)的光化学氧化反应[29].在太阳辐射强烈的条件下,M可被异构成反丁烯二酸(F).然而,在霾天气中,由于光照较弱这种异构化反应受到抑制[26].泰山气溶胶中F/M的比值与城市、高山及海洋地区的比较如图 5所示.泰山地区F/M比值为1.0±0.5,远高于城市与海洋地区,这也证实了在太阳辐射较强的高山地区,M异构成F这一反应也被加强.

|
数据来源与表 2相同 图 5 泰山地区夏季PM2.5中F/M、C9/C6和C9/Ph与其它地区的比较 Fig. 5 Comparison of mass ratios of F/M, C9/C6 and C9/Ph in Mt. Taishan with those in other regions |
C6和Ph被认为是来自人为源排放的环己烷和芳香烃化合物(例如萘)经光化学氧化生成的产物[7, 14].然而,C9是由生物源排放的不饱和脂肪酸经化学氧化形成的[5].因此,C9/C6和C9/Ph这两个比值均可用来评估人为源与生物源对SOA的影响程度.由图 5所示,泰山气溶胶中C9/C6和C9/Ph比值分别为12±7.6、1.9±1.3,高于城市、海洋与高山地区(华山除外)气溶胶的2~9倍.另外,C9占二元羧酸总浓度的相对含量(5.1%±2.5%)远高于中国北方城市的气溶胶[12],表明生物源对泰山SOA的影响很显著,而受人为源的影响相对较弱.
2.4 生物源对华山夏季二元羧酸类SOA的贡献Gly与mGly的来源与全球预算值如表 3所示,人为源与生物源产生Gly/mGly的比值分别为1:1、1:5[11].因为Gly与mGly的吸收系数是相似的,当生物源为单一排放源时[11],颗粒态Gly/mGly比值理论上应该为1:5.反之,当天然源为单一排放源时,颗粒态Gly/mGly比值应该为1:1.另外,通过华山气溶胶中Gly/mGly比值冬、夏季的对比[16],已证明这一结论的正确性.由表 3可知,泰山夏季气溶胶中mGly/Gly的比值为4.9,接近于5,表明泰山夏季Gly与mGly主要受生物源的影响.由于Gly与mGly是C2的主要前体物[6, 11],由此可推断,泰山夏季气溶胶中的C2受生物源的影响较大.
|
|
表 3 泰山夏季PM2.5样品中Gly、mGly的浓度与应用模式的估算值及华山的比较 Table 3 Comparisons with concentrations of α-dicarbonyls at Mt. Taishan samples in summer and the global budgets of atmospheric Gly and mGly and those at Mt. Huashan |
生物源挥发性有机物(BVOCs, 1 150 Tg·a-1)对大气化学具有重要的影响,主要包括异戊二烯、单萜烯(如α-/β-pinene)以及倍半萜烯(如β-caryophyllene)等.在全球范围内,BVOCs的含量约比人为源VOCs高一个数量级[32].已有研究表明短链的二元羧酸(C2~C5)主要是由夏季植物排放的BSOA的前体物(例如异戊二烯、α-/β-蒎烯、乙酸及不饱和脂肪酸)经液相氧化形成的[6].
基于3D模式,Myriokefalitakis等[33]认为C2由来自植物排放的碳氢化合物(例如异戊二烯、β-石竹烯)在云中经光化学氧化反应形成的.因此,通过分析异戊二烯、α-/β-蒎烯及β-石竹烯的氧化产物与二元羧酸类SOA的关系,讨论生物源对二元羧酸的贡献.由表 1可知,泰山夏季BSOA的示踪物中浓度最高的是异戊二烯的氧化产物,其次是α-/β-蒎烯及β-石竹烯的氧化产物. BSOA的示踪物与二元羧酸类SOA的相关性分析如图 6所示,从中可知,二元羧酸与异戊二烯的氧化产物呈很强的相关性(R2=0.73),而与α-/β-蒎烯(R2=0.54)及β-石竹烯的氧化产物(R2=0.63)的相关性相对较弱.泰山夏季异戊二烯的氧化产物浓度最高,与二元羧酸的相关性越强,因此对二元羧酸类SOA的贡献越大.另外,α-/β-蒎烯及β-石竹烯的氧化产物也与酮羧酸、α-二羰基化合物呈较强的相关性.综上所述,异戊二烯、α-/β-蒎烯及β-石竹烯的氧化产物都可作为二元羧酸类SOA的重要前体物,表明泰山夏季二元羧酸类SOA主要受当地生物源光化学氧化的影响,而非长距离传输.

|
图 6 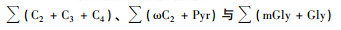 |
(1)泰山夏季PM2.5中离子浓度最高的是SO42-、其次是NO3-和NH4+. n(NH4+)/n(2×SO42+NO3-)比值为0.4,表明泰山地区气溶胶呈酸性.
(2)大气PM2.5中二元羧酸总浓度为(376±189) ng·m-3,其中C2的浓度最高,其次C3、C4、C9和Ph,与受人为污染源影响较小的偏远地区(如高山、海洋)是相似的,但与受人为影响较重的城市地区不同.
(3) C3/C4与温度间相关性较强(R2=0.67),初步确定泰山夏季二元羧酸类SOA主要来自本地源.分子组成与比值对比确定当地生物源是泰山二元羧酸类SOA的主要来源,与模式估算值的比较及与BSOA示踪物的相关性分析进而确定SOA主要来自当地生物源的光化学氧化反应,而非长距离传输.
| [1] | Wang G H, Zhou B H, Cheng C L, et al. Impact of Gobi desert dust on aerosol chemistry of Xi'an, inland China during spring 2009:differences in composition and size distribution between the urban ground surface and the mountain atmosphere[J]. Atmospheric Chemistry and Physics, 2013, 13(2) : 819–835. DOI: 10.5194/acp-13-819-2013 |
| [2] | 李璇, 聂滕, 齐珺, 等. 2013年1月北京市PM2.5区域来源解析[J]. 环境科学, 2015, 36(4) : 1148–1153. Li X, Nie T, Qi J, et al. Regional source apportionment of PM2.5 in Beijing in January 2013[J]. Environmental Science, 2015, 36(4) : 1148–1153. |
| [3] | 陈卓, 刘峻峰, 陶玮, 等. 中国地区二次有机气溶胶的时空分布特征和来源分析[J]. 环境科学, 2016, 37(8) : 2815–2822. Chen Z, Liu J F, Tao W, et al. Spatiotemporal distribution and source attribution of SOA in China[J]. Environmental Science, 2016, 37(8) : 2815–2822. |
| [4] | Hoque M, Kawamura K. Longitudinal distributions of dicarboxylic acids, ω-oxoacids, pyruvic acid, α-dicarbonyls, and fatty acids in the marine aerosols from the central Pacific including equatorial upwelling[J]. Global Biogeochemical Cycles, 2016, 30(3) : 534–548. DOI: 10.1002/gbc.v30.3 |
| [5] | Hoque M, Kawamura K, Seki O, et al. Spatial distributions of dicarboxylic acids, ω-oxoacids, pyruvic acid and α-dicarbonyls in the remote marine aerosols over the North Pacific[J]. Marine Chemistry, 2015, 172 : 1–11. DOI: 10.1016/j.marchem.2015.03.003 |
| [6] | Zhang Y L, Kawamura K, Cao F, et al. Stable carbon isotopic compositions of low-molecular-weight dicarboxylic acids, oxocarboxylic acids, α-dicarbonyls, and fatty acids:implications for atmospheric processing of organic aerosols[J]. Journal of Geophysical Research:Atmospheres, 2016, 121(7) : 3707–3717. DOI: 10.1002/2015JD024081 |
| [7] | Kawamura K, Kasukabe H, Barrie L A. Source and reaction pathways of dicarboxylic acids, ketoacids and dicarbonyls in arctic aerosols:one year of observations[J]. Atmospheric Environment, 1996, 30(10-11) : 1709–1722. DOI: 10.1016/1352-2310(95)00395-9 |
| [8] | Crahan K K, Hegg D, Covert D S, et al. An exploration of aqueous oxalic acid production in the coastal marine atmosphere[J]. Atmospheric Environment, 2004, 38(23) : 3757–3764. DOI: 10.1016/j.atmosenv.2004.04.009 |
| [9] | Sorooshian A, Lu M L, Brechtel F J, et al. On the source of organic acid aerosol layers above clouds[J]. Environmental Science & Technology, 2007, 41(13) : 4647–4654. |
| [10] | Kawamura K, Bikkina S. A review of dicarboxylic acids and related compounds in atmospheric aerosols:molecular distributions, sources and transformation[J]. Atmospheric Research, 2016, 170 : 140–160. DOI: 10.1016/j.atmosres.2015.11.018 |
| [11] | Fu T M, Jacob D J, Wittrock F, et al. Global budgets of atmospheric glyoxal and methylglyoxal, and implications for formation of secondary organic aerosols[J]. Journal of Geophysical Research:Atmospheres, 2008, 113(D15) : D15303. DOI: 10.1029/2007JD009505 |
| [12] | Cheng C L, Wang G H, Zhou B H, et al. Comparison of dicarboxylic acids and related compounds in aerosol samples collected in Xi'an, China during haze and clean periods[J]. Atmospheric Environment, 2013, 81 : 443–449. DOI: 10.1016/j.atmosenv.2013.09.013 |
| [13] | Ho K F, Cao J J, Lee S C, et al. Dicarboxylic acids, ketocarboxylic acids, and dicarbonyls in the urban atmosphere of China[J]. Journal of Geophysical Research:Atmospheres, 2007, 112(D22) : D22S27. |
| [14] | Narukawa M, Kawamura K, Takeuchi N, et al. Distribution of dicarboxylic acids and carbon isotopic compositions in aerosols from 1997 Indonesian forest fires[J]. Geophysical Research Letters, 1999, 26(20) : 3101–3104. DOI: 10.1029/1999GL010810 |
| [15] | Kundu S, Kawamura K, Lee M. Seasonal variations of diacids, ketoacids, and α-dicarbonyls in aerosols at Gosan, Jeju Island, South Korea:implications for sources, formation, and degradation during long-range transport[J]. Journal of Geophysical Research:Atmospheres, 2010, 115(D19) : D19307. DOI: 10.1029/2010JD013973 |
| [16] | Meng J J, Wang G H, Li J J, et al. Seasonal characteristics of oxalic acid and related SOA in the free troposphere of Mt. Hua, central China:implications for sources and formation mechanisms[J]. Science of the Total Environment, 2014, 493 : 1088–1097. DOI: 10.1016/j.scitotenv.2014.04.086 |
| [17] | Kawamura K, Tachibana E, Okuzawa K, et al. High abundances of water-soluble dicarboxylic acids, ketocarboxylic acids and α-dicarbonyls in the mountain aerosols over the North China Plain during wheat burning season[J]. Atmospheric Chemistry and Physics, 2013, 13(2) : 3695–3734. |
| [18] | Kawamura K, Okuzawa K, Aggarwal S G, et al. Determination of gaseous and particulate carbonyls (glycolaldehyde, hydroxyacetone, glyoxal, methylglyoxal, nonanal and decanal) in the atmosphere at Mt. Tai[J]. Atmospheric Chemistry and Physics, 2013, 13(10) : 5369–5380. DOI: 10.5194/acp-13-5369-2013 |
| [19] | Wang G H, Li J J, Cheng C L, et al. Observation of atmospheric aerosols at Mt. Tai in central and east China during spring 2009-Part 1:EC, OC and inorganic ions[J]. Atmospheric Chemistry and Physics, 2011, 11(9) : 4221–4235. DOI: 10.5194/acp-11-4221-2011 |
| [20] | Wang G H, Li J J, Cheng C L, et al. Observation of atmospheric aerosols at Mt. Hua and Mt. Tai in central and east China during spring 2009-Part 2:impact of dust storm on organic aerosol composition and size distribution[J]. Atmospheric Chemistry and Physics, 2012, 12(9) : 4065–4080. DOI: 10.5194/acp-12-4065-2012 |
| [21] | Fu P Q, Kawamura K, Kanaya Y, et al. Contributions of biogenic volatile organic compounds to the formation of secondary organic aerosols over Mt. Tai, Central East China[J]. Atmospheric Environment, 2010, 44(38) : 4817–4826. DOI: 10.1016/j.atmosenv.2010.08.040 |
| [22] | Cao J J, Lee S C, Chow J C, et al. Spatial and seasonal distributions of carbonaceous aerosols over China[J]. Journal of Geophysical Research:Atmospheres, 2007, 112(D22) : D22S11. |
| [23] | Meng J J, Wang G H, Li J J, et al. Atmospheric oxalic acid and related secondary organic aerosols in Qinghai Lake, a continental background site in Tibet Plateau[J]. Atmospheric Environment, 2013, 79 : 582–589. DOI: 10.1016/j.atmosenv.2013.07.024 |
| [24] | Xu H, Wang Y, Wen T, et al. Characteristics and source apportionment of atmospheric aerosols at the summit of Mount Tai during summertime[J]. Atmospheric Chemistry and Physics Discussions, 2009, 9(4) : 16361–16379. DOI: 10.5194/acpd-9-16361-2009 |
| [25] | Surratt J D, Chan A W H, Eddingsaas N C, et al. Reactive intermediates revealed in secondary organic aerosol formation from isoprene[J]. Proceedings of the National Academy of Sciences of the United States of America, 2010, 107(15) : 6640–6645. DOI: 10.1073/pnas.0911114107 |
| [26] | Hegde P, Kawamura K. Seasonal variations of water-soluble organic carbon, dicarboxylic acids, ketocarboxylic acids, and α-dicarbonyls in Central Himalayan aerosols[J]. Atmospheric Chemistry and Physics, 2012, 12(14) : 6645–6665. DOI: 10.5194/acp-12-6645-2012 |
| [27] | He N N, Kawamura K, Okuzawa K, et al. Diurnal and temporal variations of water-soluble dicarboxylic acids and related compounds in aerosols from the northern vicinity of Beijing:implication for photochemical aging during atmospheric transport[J]. Science of the Total Environment, 2014, 499 : 154–165. DOI: 10.1016/j.scitotenv.2014.08.050 |
| [28] | Pavuluri C M, Kawamura K, Swaminathan T. Water-soluble organic carbon, dicarboxylic acids, ketoacids, and α-dicarbonyls in the tropical Indian aerosols[J]. Journal of Geophysical Research:Atmospheres, 2010, 115(D11) : D11302. DOI: 10.1029/2009JD012661 |
| [29] | Aggarwal S G, Kawamura K. Molecular distributions and stable carbon isotopic compositions of dicarboxylic acids and related compounds in aerosols from Sapporo, Japan:implications for photochemical aging during long-range atmospheric transport[J]. Journal of Geophysical Research:Atmospheres, 2008, 113(D14) : D14301. DOI: 10.1029/2007JD009365 |
| [30] | Wang H B, Kawamura K, Yamazaki K. Water-Soluble dicarboxylic acids, ketoacids and dicarbonyls in the atmospheric aerosols over the southern ocean and western pacific ocean[J]. Journal of Atmospheric Chemistry, 2006, 53(1) : 43–61. DOI: 10.1007/s10874-006-1479-4 |
| [31] | Jung J, Tsatsral B, Kim Y J, et al. Organic and inorganic aerosol compositions in Ulaanbaatar, Mongolia, during the cold winter of 2007 to 2008:Dicarboxylic acids, ketocarboxylic acids, and α-dicarbonyls[J]. Journal of Geophysical Research:Atmospheres, 2010, 115(D22) : D22203. DOI: 10.1029/2010JD014339 |
| [32] | Guenther A, Karl T, Harley P, et al. Estimates of global terrestrial isoprene emissions using MEGAN (Model of Emissions of Gases and Aerosols from Nature)[J]. Atmospheric Chemistry and Physics, 2006, 6(11) : 3181–3210. DOI: 10.5194/acp-6-3181-2006 |
| [33] | Myriokefalitakis S, Tsigaridis K, Mihalopoulos N, et al. In-cloud oxalate formation in the global troposphere:a 3-D modeling study[J]. Atmospheric Chemistry and Physics, 2011, 11(12) : 5761–5782. DOI: 10.5194/acp-11-5761-2011 |
| [34] | Mochida M, Kawabata A, Kawamura K, et al. Seasonal variation and origins of dicarboxylic acids in the marine atmosphere over the western North Pacific[J]. Journal of Geophysical Research:Atmospheres, 2003, 108(D6) : 4193. DOI: 10.1029/2002JD002355 |