 2017, Vol. 38
2017, Vol. 38

2. 重庆大学低碳绿色建筑国际联合研究中心, 重庆 400045;
3. 中国城市规划设计研究院西部分院, 重庆 400021;
4. 石河子大学水利建筑工程学院, 石河子 832003
 , ZHANG Shuai1 , ZHOU Yu3 , LI Kun1 , ZHOU Wei1 , LI Peng-yu1 , YANG Guang4 , SUN Zhi-hua4 , ZHENG Huai-li1,2
, ZHANG Shuai1 , ZHOU Yu3 , LI Kun1 , ZHOU Wei1 , LI Peng-yu1 , YANG Guang4 , SUN Zhi-hua4 , ZHENG Huai-li1,2
2. National Centre for International Research of Low-carbon and Green Buildings, Chongqing University, Chongqing 400045, China;
3. West Branch, China Academy of Urban Planning & Design, Chongqing 400021, China;
4. College of Water Conservancy and Architectural Engineering, Shihezi University, Shihezi 832003, China
作为一种硝基芳香族化合物,硝基苯中硝基为吸电子基团从而难以被微生物或其他物质氧化或还原,是一种典型的难化学降解的有毒有机污染物.硝基苯具有致癌毒性[1],被广泛用于农药、 染料、 炸药的生产中[2, 3].它也可以用来作为抛光工业溶剂等.即使在低浓度下,硝基苯也存在着较高的环境风险.硝基苯可通过呼吸作用及皮肤侵入人体影响中枢神经系统,从而产生疲劳、 头痛、 眩晕、 呕吐、 全身无力、 无意识、 昏迷等症状[4].硝基化合物的浓度在我国排放废水中有严格的要求,如在《污水综合排放标准》 (GB 897821996)中规定硝基化合物的浓度不得超过5 mg·L-1.因此,将硝基苯分解为无毒无害物质非常必要.目前,硝基苯的处理方法大致可分三类:生物方法、 物理方法和化学方法[5~7].这些方法各有优缺点,但很难达到理想的去除效果.
臭氧是一种具有刺激性特殊气味的不稳定气体,其氧化能力极强(氧化还原电位2.075 V,仅次于氟),是一种高效的无二次污染的氧化剂[8].将活性炭与臭氧技术联用去除水中色度时两者能产生很好的耦合效果[9].即使少量的活性炭也会使得臭氧在水中分解并产生羟基自由基[10].虽然臭氧的分解能力与活性炭表面活性点的数量有关,但是在活性炭存在的情况下,臭氧的分解速率和自由基生成速率依然比单独臭氧快三倍.因此在臭氧化中活性炭可以被认为是通过一种自由基路径的催化剂.此外,Jans等[10]认为臭氧分解发生的活性炭表面并产生羟基自由基,羟基自由基再与水中的有机分子发生反应.尽管将活性炭用于催化臭氧化可以提高臭氧的利用效率并加强污染物的矿化,但是在不同文献中随着反应时间的加长活性炭都表现出了部分失活现象[11, 12]. Sánchez-Polo等[12]将活性炭在通入气相臭氧并重复120次后发现其表面酸性含氧官能团增多,表面积减少.
本研究以电增强活性炭纤维-臭氧体系去除水中硝基苯,分析其降解效率,探索电流强度、 电解质种类及其浓度等实验条件对电化学/臭氧体系的影响,并探讨了其降解机制.
1 材料与方法 1.1 材料及仪器硝基苯(纯度>99%)购自成都市科龙化工试剂厂,实验中所用到的硝酸钠、 氯化钠、 硫酸钠、 碘化钾、 草酸钛钾等均为分析纯.实验中所用的溶液均采用超纯水(18.2 MΩ·cm)配制.
高效液相色谱仪(Waters2487),美国Waters公司.磁力搅拌器(MS300),上海般特仪器有限公司. pH计(PHS/3C),上海大普仪器有限公司.臭氧发生器(MS6610),东莞华仪仪表科技有限公司.直流稳压电源(WYJ),上海兴林稳压电源有限公司.气流计(MS6610),东莞华仪仪表科技有限公司.超纯水制备仪(GWA. UN),美国Waters公司.
本实验在一个圆柱形电化学/臭氧反应器中进行(如图 1).该反应器高15 cm,直径为7 cm.将配制完成的500 mL浓度为12 mg·L-1的硝基苯溶液加入反应器中,反应器的底部为磁力搅拌器,反应器中转子的转速为1 200 r·min-1.将活性炭纤维用钛丝捆绑至钛金属板固定,活性炭纤维的有效面积与钛镀铂金属阴/阳极相同,尺寸为3.5 mm×10 mm,阴阳极的间距为1 cm.当电化学技术与臭氧技术联用时,首先打开直流电源,再打开臭氧发生器.当实验中需要用到电场时,在无特殊说明的情况下,向反应液中添加浓度为0.05 mmol·L-1的硫酸钠电解质.此外,实验中一般情况下臭氧的浓度为8.33 mg·L-1,电流的强度为100 mA.

|
图 1 反应器示意 Fig. 1 Schematic diagram of the reactor |
实验中硝基苯的初始浓度及反应后的浓度通过高效液相色谱(HPLC)测定.分析条件为:Waters2487液相色谱,紫外检测器.色谱柱为反相C18柱(COSMOSIL 3C18-MS-Ⅱ,5 μmol·L-1,4.6 mm×150 mm,Nacalai Tesque,Inc.,Japan),测试时色谱柱温度为25℃; 流动相为体积比55∶40∶5的水/甲醇/乙腈的混合液,流速为1mL·min-1,检测波长为254 nm,进样量为10 μL.
液相过氧化氢浓度利用草酸钛钾法进行测定[13]. 准确量取136 mL浓硫酸缓慢加入150 mL的超纯水中,向此溶液中加入17.7 g草酸钛钾后用超纯水定容至0.5 L,得到0.1mol·L-1的草酸钛钾溶液,取5 mL水样加入25 mL的容量瓶后用0.1 mol·L-1的草酸钛钾溶液标定,反应10 min后在λ=400 nm处测其吸光度.
2 结果与讨论 2.1 阴极电场对臭氧/活性炭纤维体系的促进效果考察了电增强活性炭纤维-臭氧、 活性炭纤维-臭氧、 电增强活性炭纤维、 电化学-臭氧、 钛阴极电解、 臭氧对目标污染物硝基苯去除效果.硝基苯初始浓度为12 mg·L-1,臭氧浓度为8.33 mg·L-1,电流的强度为100 mA,硫酸钠电解质添加浓度为0.05 mmol·L-1,反应时间为20 min,所用的活性炭纤维大小形状相同.从图 2中可以看出单独的臭氧和电解对硝基苯的去除效率很低,20 min降解率分别为31.10%和23.54%.将活性炭纤维用于臭氧的催化过程时,硝基苯的降解速率得到明显提升,其降解率为80.92%.在活性炭纤维-臭氧体系中将活性炭纤维表面施加一个阴极电场时,硝基苯降解率达到91.19%,说明电解与活性炭纤维催化臭氧化联用时的降解效率不是简单地对两种技术的叠加,将阴极电场施加在活性炭纤维表面时还会产生一定的耦合效果.而在此体系中,将活性炭纤维运用于电解反应中时其反应速率也远不及耦合体系的效果,其去除率为58.44%. 2005年时Kishimoto等[14]就提出将电解与臭氧结合时观测到较强的耦合效果,但是在此体系(电化学-臭氧)硝基苯的去除效率也仅有71.42%.以上说明将阴极电场运用于活性炭纤维-臭氧体系能显著提升硝基苯的去除效率.

|
图 2 不同工艺对目标污染物硝基苯去除效果 Fig. 2 Removal of NB by different processes |
为了确定硝基苯在电增强活性炭纤维-臭氧体系中的降解机制,分别测试了活性炭纤维在电解和电化学/臭氧体系中过氧化氢的浓度.如图 3(a)所示,当活性炭纤维作为阴极电解硝基苯溶液时,反应液中过氧化氢的浓度随时间呈线性上升,这说明电解反应产生的过氧化氢与硝基苯的反应活性很低[15].而当体系中通入臭氧后,反应进行到第8 min时过氧化氢的浓度增幅变缓,这说明加入的臭氧与过氧化氢发生了反应并产生羟基自由基[16],羟基自由基使得电增强活性炭纤维-臭氧体系中的硝基苯氧化.有研究者[11]在考察活性炭对臭氧分解常数的影响时发现,水中活性炭与臭氧接触后的第1~2 min内产生了大约2~3 μmol·L-1的过氧化氢,此后过氧化氢的浓度缓慢下降.当向溶液中加入相似浓度的过氧化氢时,臭氧的分解常数并未产生显著区别.这说明活性炭纤维对臭氧的促进作用主要是活性炭纤维释放的低浓度过氧化氢产生的.但是,当在活性炭纤维上施加阴极电场时,如图 3(b)所示,5 min时反应液中过氧化氢浓度约为36~60 μmol·L-1并快速地增加.因此当在活性炭纤维-臭氧体系中对活性炭纤维施加阴极电场时,更多的过氧化氢与臭氧发生反应而使得臭氧分解形成羟基自由基的效率增加.因此,在电增强活性炭纤维-臭氧体系中,阴极电场的增强作用主要是通过在活性炭纤维阴极表面生成的更多的过氧化氢等触发臭氧转变为更多的羟基自由基,而非对硝基苯的直接电解氧化或还原及臭氧对硝基苯的降解. 当两种体系中不添加硝基苯时,经过测定过氧化氢浓度与添加硝基苯并无明显差异,说明硝基苯并不能与过氧化氢发生反应.

|
图 3 活性炭纤维在电解体系及电化学/臭氧体系中作为阴极时过氧化氢浓度的变化 Fig. 3 Change of the concentration of hydrogen peroxide when ACF was used as cathode in electrolysis and E-peroxone |
当在活性炭纤维上施加阴极电场时,首先应该考虑的是电流强度对体系的影响.如图 4(a)所示,实验分别选取50、 100、 150及200 mA的电流强度研究其对体系的影响情况,其中R2为反应相关系数.从中可知,硝基苯的降解过程符合准一级动力学模型.当电流强度从50 mA增加到200 mA时,硝基苯降解速率分别为0.118 5、 0.119 7、 0.118 6、 0.110 1 min-1,整体上电流强度对体系降解率的影响较小.这一方面是因为开路时活性炭纤维就能快速地吸附硝基苯,而在活性炭纤维上施加电场对硝基苯的吸附速率影响不大[17]; 另一方面,在活性炭纤维上施加阴极电场时,活性炭纤维表面有过氧化氢产生[式(1)],虽然增加电流强度使得溶液中过氧化氢的产量增加,从而与臭氧发生反应[式(2)]生成更多的羟基自由基加速硝基苯的降解[18],但是当电流增大时,相应的阴阳极电压也同样增大,生成的过量过氧化氢难以直接氧化硝基苯,而是使得更多的过氧化氢在阳极或溶液中发生分解反应[19, 20] [式(3)及式(4)].
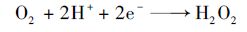
|
(1) |
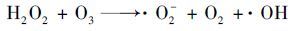
|
(2) |

|
(3) |
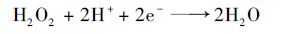
|
(4) |

|
图 4 中文标题 Fig. 4 电流强度及臭氧浓度对电增强活性炭纤维-臭氧体系的影响 |
此外,由于臭氧在水中的溶解度很低,气相臭氧扩散至液相臭氧的过程一直是制约臭氧转变为羟基自由基反应速率的一个重要因素[14].当体系中臭氧的量不足时,进一步提高过氧化氢的生成量并不会使得羟基自由基的量增加.如图 4(b)所示,当气相中的臭氧浓度从2.08 mg·L-1增加到8.33 mg·L-1时,硝基苯的降解速率从0.073 1 min-1增加到0.119 8 min-1.提高气相中臭氧浓度会使得臭氧从气相转移至液相的速率上升,更多的臭氧与过氧化氢反应产生羟基自由基[21].此外,根据Kishimoto等[22]提出的反应机制,液相中的臭氧在阴极得到电子生成羟基自由基,因此提高液相中臭氧浓度会使得更多的臭氧发生反应生成强氧化性的羟基自由基而使得硝基苯的降解速率上升.
2.2.2 溶液初始pH值对硝基苯降解效果的影响图 5(a)为溶液初始pH值对活性炭纤维催化臭氧的影响,反应符合准一级动力学模型.反应条件为气相臭氧浓度8 mg·L-1,硝基苯初始浓度为12 mg·L-1,在酸性pH的条件下,活性炭纤维催化臭氧化的能力最弱,当溶液初始pH值升高至5时活性炭纤维催化臭氧体系的对硝基苯的降解速率从0.068 9 min-1增加到0.116 3 min-1.当溶液pH值呈中性或碱性时,活性炭纤维催化臭氧的能力得到显著的提升.但是将阴极电场施加于活性炭纤维时反应初始pH值对硝基苯降解速率的影响十分微弱,无论是在酸性、 中性或碱性条件下,对硝基苯的降解速率都比较高[见图 5(b),电流强度为100 mA,其余条件与5(a)相同].

|
图 5 溶液初始 pH值对活性炭纤维催化臭氧化和电增强活性炭纤维-臭氧体系的影响 Fig. 5 Effect of initial pH value on catalytic ozonation and the electrochemically enhanced ACF/O3 system |
由于具有共振结构,臭氧分子可以作为偶极反应的亲电或亲核剂.因此臭氧分子在水中的反应活性较高,其半衰期随着溶液pH值及温度等变化而改变,大约有几秒至几分钟.通常情况下,臭氧分子在纯水溶液中发生以下链式反应[23]:

|
(5) |
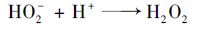
|
(6) |

|
(7) |
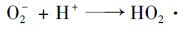
|
(8) |

|
(9) |

|
(10) |
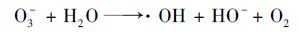
|
(11) |

|
(12) |
以上反应式说明氢氧根离子在臭氧分解中起着十分重要的作用,在溶液pH呈碱性时,更多的羟基自由基是通过氢氧根离子触发链式反应生成的.当活性炭纤维在含有硝基苯的水中催化臭氧时,硝基苯及臭氧吸附至活性炭纤维表面发生反应.在酸性环境(pH 2~6)时,硝基苯被快速地吸附至活性炭纤维表面,水中臭氧与吸附至活性炭纤维表面的硝基苯发生反应.但是当溶液pH值进一步上升呈碱性(pH 7~9)时,水中的氢氧根离子也快速地被吸附至活性炭纤维表面,如前所述,氢氧根离子会使得臭氧快速地转换为羟基自由基从而导致硝基苯的去除[24].此外,当溶液的pH呈碱性时,反应液中发生的静电反应也起着重要的作用.活性炭纤维表面可以随着溶液pH的变化而带正电或负电,当活性炭纤维的pHpzc>pH时,活性炭纤维表面带正电,反之则带负电.带正电的活性炭纤维可以通过静电作用吸引水中的氢氧根离子,因此增加反应液的pH值有助于活性炭纤维催化臭氧转变成为羟基自由基.
2.2.3 电解质对硝基苯降解效果的影响为了研究活性炭纤维阴极/臭氧体系中电解质的影响,选用0.05mol·L-1的硫酸钠、 硝酸钠及氯化钠作为反应体系的电解质.如图 6(a)所示,反应符合准一级动力学反应模型.当反应体系中加入硫酸钠及硝酸钠作为电解质时,反应体系对硝基苯的降解速率很大.而使用氯化钠作为反应电解质时,硝基苯的降解受到了抑制,甚至低于单独活性炭纤维催化臭氧化降解硝基苯的效率.为了与活性炭纤维阴极/臭氧体系作比较,在活性炭纤维-臭氧体系中加入相同种类及数量的无机盐,如图 6(b)所示,其大致的反应趋势与活性炭纤维阴极/臭氧体系相同,即不同盐的降解速率为0.05mol·L-1 Na2SO4>0.05mol·L-1 NaNO3>0.05mol·L-1NaCl.此外,与未添加任何无机盐相比,在活性炭纤维-臭氧中添加无机盐使得体系的降解效率下降.

|
图 6 电解质对活性炭纤维阴极/臭氧以及活性炭纤维-臭氧体系的影响 Fig. 6 Effect of electrolyte on catalytic ozonation and the electrochemically enhanced ACF/O3 system |
向纯水溶液中添加无机盐时,有机污染物在水中的溶解度会发生改变[25].因此,在活性炭纤维-臭氧体系中,向反应液添加无机盐会使得硝基苯从液相扩散至活性炭纤维表面的效率下降,从而使得硝基苯的降解效率发生改变.而在活性炭纤维阴极/臭氧体系中,为了增加电流传输效率,必须向反应液中添加一定量的无机盐.在活性炭纤维阴极/臭氧体系中氯化钠的存在会使得反应体系的效率明显下降,甚至低于活性炭纤维催化臭氧对硝基苯的去除.在活性炭阴极/臭氧体系降解硝基苯的过程中,在开始反应的1~2 min内反应液由无色变为黄色,并随着反应时间的增加其反应液的色度逐渐上升.
这可能是因为氯离子与硝基苯的中间产物发生了一系列的反应并产生了聚合物,这些聚合物聚集在活性炭纤维的表面阻塞了活性炭纤维对硝基苯的吸附,从而影响了体系对硝基苯的去除效率.这一现象在其他电化学/臭氧方面的文献也有发现,如Qiu 等[26]利用电化学/臭氧体系降解对硝基苯酚时,发现提高氯化钠的浓度反而会使得有机物的矿化效率下降.而当使用硫酸钠作为电解质时,反应体系中的活性物质(如·OH)会使得溶液中的硫酸根发生[式(13)]反应,是一种强氧化剂而使得硝基苯被加速氧化.

|
(13) |
活性炭与臭氧结合降解有机污染物的机制已经被很多学者研究.活性炭主要通过促进臭氧分解生成羟基自由基,如Alvárez等[27]认为臭氧与活性炭纤维表面的官能团(主要是含氧官能团)反应生成了少量过氧化氢,过氧化氢扩散至反应液中,水中的臭氧在过氧化氢的促发作用下生成羟基自由基.
由于实验室条件限制,无法直接检测羟基自由基的生成,只能通过自由基捕获剂间接检测. 由于叔丁醇与羟基自由基的反应常数大[约为6×108 L·(mol·s)-1],而与臭氧的反应常数很小[约为3×10-3 L·(mol·s)-1],因此常被用作检测反应液中羟基自由基的生成. 因此,当向反应初始液中添加叔丁醇时,臭氧与活性炭反应生成的羟基自由基被叔丁醇淬灭,从而导致水中目标污染物的降解效率下降.当向反应体系中加入1 mmol·L-1的叔丁醇时,硝基苯的降解效率在20 min时由91.19%下降为56.65%,去除率大大降低,这说明反应体系中有羟基自由基产生.
2.4 活性炭纤维表面性质的比较为了进一步对活性炭纤维表面进行观察,对原始活性炭纤维、 臭氧反应后的活性炭纤维及在电场保护下与臭氧反应后的活性炭纤维进行了电镜扫描,臭氧与活性炭纤维反应进行12 h,且将臭氧浓度调整为0.83 g·L-1,在相同条件下,另外一块活性炭纤维在阴极电场的保护下与臭氧进行反应.如图 7所示,原始活性炭纤维表面光滑,而经过臭氧反应后的活性炭纤维表面被臭氧腐蚀,表面凹凸不平.而在阴极电场保护下的活性炭纤维未受到臭氧的腐蚀,表面依然光滑.

|
图 7 原始活性炭纤维、 与臭氧反应后的活性炭纤维和在阴极电场保护下与臭氧反应的活性炭纤维的扫描电镜图 Fig. 7 SEM images of the texture of virgin,O3 reacted,E-peroxone reacted ACFs |
以上结果说明水中臭氧与活性炭纤维的微孔壁发生反应使得活性炭纤维的微孔结构被破坏,从而导致活性炭纤维表面积下降.活性炭纤维的表面积主要通过测试活性炭纤维对氮气的吸附量,因此臭氧与活性炭纤维表面发生反应后在微孔入口处形成的含氧官能团可能会阻碍氮气的进入[28].此外,虽然1次反应20 min时硝基苯的去除率为95%左右,但是其反应中间产物还可能占据活性炭纤维表面的吸附点,因此硝基苯中间产物的不完全降解也可能造成活性炭纤维的比表面减少.在活性炭纤维上施加阴极电场时使得活性炭纤维成为一种电催化剂,提高了反应体系中有机污染物的降解效率[29]. Ania等[30]发现阴极电场对活性炭具有保护作用,当水中没有其他吸附质存在的情况下,在活性炭表面施加阴极电场不会造成结构性质或化学性质的变化.在活性炭阴极的石墨层存在着一层“网状”电子,当含氧活性物质(如·OH)靠近时,活性物质获得电子变成氧化性较弱或者无氧化性的物质.如臭氧与羟基自由基发生下反应:

|
(14) |

|
(15) |
因此,当水中的强氧化性物质如臭氧、 羟基自由基等接收活性炭阴极外表面的电子后,生成的弱氧化性物质难以对活性炭纤维的物理或化学性质造成影响.
此外,Berenquer等[31]研究了阴极电场对活性炭的影响,发现在反应液中通入气体有利于活性炭表面积的增加,且电产生的过氧化氢对活性炭表面不会造成显著地影响.本实验中未观察到活性炭纤维表面或微孔面积的增加,这可能是因为反应时间太短(20 min)的缘故,水中硝基苯及其中间产物吸附在活性炭纤维的微孔或中孔入口处,使得活性炭纤维的表面积、 微孔面积、 微孔体积及总体积都发生了微弱的下降.与此同时,电产生大量的过氧化氢扩散至水中与臭氧发生反应生成羟基自由基.由于羟基自由基的强氧化性(~2.8 V),因此存在时间很短(约为1×10-9 s),造成其扩散距离很小[11],使得达到活性炭纤维表面的臭氧、 羟基自由基或其他活性物质减少,从而减小了臭氧氧化活性炭纤维表面的可能性,避免了臭氧对活性炭纤维表面官能团或结构的破坏.
3 结论(1) 与活性炭纤维-臭氧体系比较,阴极电场增强活性炭纤维-臭氧体系对硝基苯的去除效率显著提升.
(2) 阴极电场增强活性炭纤维-臭氧体系中电流强度对体系的影响不显著.但是当提高或降低气相中臭氧浓度时,水中硝基苯的去除效率相应地升高或下降.
(3) 反应溶液的初始pH值对活性炭纤维催化臭氧体系的影响较大.在一定范围内,pH值越大活性炭纤维的催化能力越强.但反应溶液的初始pH值对电增强活性炭纤维-臭氧体系的影响不明显,在酸性或碱性条件下对硝基苯都保持了较高的去除效率.
(4) 水中无机盐如硫酸钠、 硝酸钠及氯化钠的存在会抑制活性炭纤维催化臭氧的能力,其中氯化钠的抑制效果最强,硫酸钠的抑制效果最弱.在电增强活性炭纤维-臭氧体系降解中,以硝酸钠电解质为基准,硫酸钠电解质对体系降解效率具有促进作用,而氯化钠电解质对体系有抑制效果.
(5) 单独臭氧对活性炭纤维具有破坏作用,降低了活性炭纤维对反应的促进效果,外加阴极电场时,不仅活性炭纤维对有机物的去除效果显著提升,而且保证了活性炭纤维不被臭氧所破坏.
| [1] | 孙猛, 赵勇胜, 董军, 等. 地下环境中Fe2+对硝基苯的还原衰减作用模拟研究[J]. 环境科学, 2011, 32(5) : 1372–1376. Sun M, Zhao Y S, Dong J, et al. Simulation study on reductive attenuation of nitrobenzene by Fe2+ in subsurface environment[J]. Environmental Science, 2011, 32(5) : 1372–1376. |
| [2] | 赵雷, 马军, 孙志忠, 等. 蜂窝陶瓷催化臭氧化降解水中微量硝基苯的动力学研究[J]. 环境科学, 2007, 28(1) : 102–107. Zhao L, Ma J, Sun Z Z, et al. Kinetics of ceramic honeycomb-catalyzed ozonation for the degradation of trace nitrobenzene in aqueous solution[J]. Environmental Science, 2007, 28(1) : 102–107. |
| [3] | Mu Y, Rozendal R A, Rabaey K, et al. Nitrobenzene removal in bioelectrochemical systems[J]. Environmental Science & Technology, 2009, 43(22) : 8690–8695. |
| [4] | 杨忆新, 马军, 秦庆东, 等. 臭氧/纳米TiO2催化氧化去除水中微量硝基苯的研究[J]. 环境科学, 2006, 27(10) : 2028–2034. Yang Y X, Ma J, Qin Q D, et al. Degradation of trace nitrobenzene by nanosized TiO2 catalyzed ozonation[J]. Environmental Science, 2006, 27(10) : 2028–2034. |
| [5] | Majumder P S, Gupta S K. Hybrid reactor for priority pollutant nitrobenzene removal[J]. Water Research, 2003, 37(18) : 4331–4336. DOI: 10.1016/S0043-1354(03)00436-6 |
| [6] | 韦朝海, 陈传好, 王刚, 等. Fenton试剂催化氧化降解含硝基苯废水的特性[J]. 环境科学, 2001, 22(5) : 60–64. Wei C H, Chen C H, Wang G, et al. Characteristics of nitrobenzene containing wastewater catalytic oxidation degradation by Fenton reagent[J]. Environmental Science, 2001, 22(5) : 60–64. |
| [7] | Rauthula M S, Srivastava V C. Studies on adsorption/desorption of nitrobenzene and humic acid onto/from activated carbon[J]. Chemical Engineering Journal, 2011, 168(1) : 35–43. DOI: 10.1016/j.cej.2010.12.026 |
| [8] | 王佳裕, 戴启洲, 鱼杰, 等. 活性炭催化臭氧氧化扑热息痛的机制研究[J]. 环境科学, 2013, 34(4) : 1402–1410. Wang J Y, Dai Q Z, Yu J, et al. Mechanism of catalytic ozonation for the degradation of paracetamol by activated carbon[J]. Environmental Science, 2013, 34(4) : 1402–1410. |
| [9] | Mckay G, McAleavey G. Ozonation and carbon adsorption in a three phase fluidized bed for color removal from peat water[J]. Chemical Engineering Research & Design, 1988, 66(6) : 531–536. |
| [10] | Jans U, Hoigné J. Activated carbon and carbon black catalyzed transformation of aqueous ozone into OH-radicals[J]. Ozone:Science & Engineering, 1998, 20(1) : 67–90. |
| [11] | Faria P C C, Órfão J J M, Pereira M F R, et al. Ozone decomposition in water catalyzed by activated carbon:influence of chemical and textural properties[J]. Industrial & Engineering Chemistry Research, 2006, 45(8) : 2715–2721. |
| [12] | Sánchez-Polo M, von Gunten U, Rivera-Utrilla J. Efficiency of activated carbon to transform ozone into·OH radicals:influence of operational parameters[J]. Water Research, 2005, 39(14) : 3189–3198. DOI: 10.1016/j.watres.2005.05.026 |
| [13] | Sellers R M. Spectrophotometric determination of hydrogen peroxide using potassium titanium(Ⅳ) oxalate[J]. Analyst, 1980, 105(1255) : 950–954. DOI: 10.1039/an9800500950 |
| [14] | Kishimoto N, Morita Y, Tsuno H, et al. Advanced oxidation effect of ozonation combined with electrolysis[J]. Water Research, 2005, 39(19) : 4661–4672. DOI: 10.1016/j.watres.2005.09.001 |
| [15] | Li X, Wang Y J, Yuan S, et al. Degradation of the anti-inflammatory drug ibuprofen by electro-peroxone process[J]. Water Research, 2014, 63 : 81–93. DOI: 10.1016/j.watres.2014.06.009 |
| [16] | Wang H J, Yuan S, Zhan J H, et al. Mechanisms of enhanced total organic carbon elimination from oxalic acid solutions by electro-peroxone process[J]. Water Research, 2015, 80 : 20–29. DOI: 10.1016/j.watres.2015.05.024 |
| [17] | Han Y H, Quan X, Zhao H M, et al. Kinetics of enhanced adsorption by polarization for organic pollutants on activated carbon fiber[J]. Frontiers of Environmental Science & Engineering in China, 2007, 1(1) : 83–88. |
| [18] | Bakheet B, Yuan S, Li Z X, et al. Electro-peroxone treatment of Orange Ⅱ dye wastewater[J]. Water Research, 2013, 47(16) : 6234–6243. DOI: 10.1016/j.watres.2013.07.042 |
| [19] | Wang C T, Chou W L, Chung M H, et al. COD removal from real dyeing wastewater by electro-Fenton technology using an activated carbon fiber cathode[J]. Desalination, 2010, 253(1-3) : 129–134. DOI: 10.1016/j.desal.2009.11.020 |
| [20] | Wang A M, Qu J H, Ru J, et al. Mineralization of an azo dye Acid Red 14 by electro-Fenton's reagent using an activated carbon fiber cathode[J]. Dyes and Pigments, 2005, 65(3) : 227–233. DOI: 10.1016/j.dyepig.2004.07.019 |
| [21] | Li Z X, Yuan S, Qiu C C, et al. Effective degradation of refractory organic pollutants in landfill leachate by electro-peroxone treatment[J]. Electrochimica Acta, 2013, 102 : 174–182. DOI: 10.1016/j.electacta.2013.04.034 |
| [22] | Kishimoto N, Yasuda Y, Mizutani H, et al. Applicability of ozonation combined with electrolysis to 1,4-dioxane removal from wastewater containing radical scavengers[J]. Ozone:Science & Engineering, 2007, 29(1) : 13–22. |
| [23] | Zhao L, Ma J, Sun Z Z, et al. Catalytic ozonation for the degradation of nitrobenzene in aqueous solution by ceramic honeycomb-supported manganese[J]. Applied Catalysis B:Environmental, 2008, 83(3-4) : 256–264. DOI: 10.1016/j.apcatb.2008.02.009 |
| [24] | Beltrán F J, Rivas J, álvarez P, et al. Kinetics of heterogeneous catalytic ozone decomposition in water on an activated carbon[J]. Ozone:Science & Engineering, 2002, 24(4) : 227–237. |
| [25] | Xie W H, Shi W Y, Mackay D. A review of the effect of salts on the solubility of organic compounds in seawater[J]. Marine Environmental Research, 1997, 44(4) : 429–444. DOI: 10.1016/S0141-1136(97)00017-2 |
| [26] | Qiu C C, Yuan S, Li X, et al. Investigation of the synergistic effects for p-nitrophenol mineralization by a combined process of ozonation and electrolysis using a boron-doped diamond anode[J]. Journal of Hazardous Materials, 2014, 280 : 644–653. DOI: 10.1016/j.jhazmat.2014.09.001 |
| [27] | Alvárez P M, García-Araya J F, Beltrán F J, et al. The influence of various factors on aqueous ozone decomposition by granular activated carbons and the development of a mechanistic approach[J]. Carbon, 2006, 44(14) : 3102–3112. DOI: 10.1016/j.carbon.2006.03.016 |
| [28] | Shang K F, Li J, Lu N, et al. Ozonation of p-Nitrophenol adsorbed on activated carbon fiber (ACF) and the change of textural and chemical characteristics of ACF[J]. Ozone:Science & Engineering, 2015, 37(2) : 178–185. |
| [29] | Zhang X K, Zhou Y, Zhao C, et al. Electric field induced activated carbon fiber (ACF) cathode transition from an initiator/a promoter into an electrocatalyst in ozonation process[J]. Chemical Engineering Journal, 2016, 304 : 129–133. DOI: 10.1016/j.cej.2016.06.079 |
| [30] | Ania C O, Béguin F. Electrochemical regeneration of activated carbon cloth exhausted with bentazone[J]. Environmental Science & Technology, 2008, 42(12) : 4500–4506. |
| [31] | Berenquer R, Marco-Lozar J P, Quijada C, et al. Effect of electrochemical treatments on the surface chemistry of activated carbon[J]. Carbon, 2009, 47(4) : 1018–1027. DOI: 10.1016/j.carbon.2008.12.022 |