 2015,Vol. 36
2015,Vol. 36

2. 青岛科技大学环境与安全工程学院,青岛 266042;
3. 中国科学院大学资源与环境学院,北京 100049
 ,CUI Bing-jian1,3,JIN De-cai1
,CUI Bing-jian1,3,JIN De-cai1 ,WU Shang-hua1,YANG Bo2,DENG Ye1,ZHUANG Guo-qiang1,ZHUANG Xu-liang1
,WU Shang-hua1,YANG Bo2,DENG Ye1,ZHUANG Guo-qiang1,ZHUANG Xu-liang1
2. College of Environment and Safety Engineering,Qingdao University of Science and Technology,Qingdao 266042,China;
3. College of Resources and Environment,University of Chinese Academy of Sciences,Beijing 100049,China
我国水资源的空间分布极不均匀,水资源供需矛盾突出,经济社会的发展对用水需求已大大超过了现有水资源的承载能力. 我国总用水量中,农业灌溉用水占70%,水资源危机已成为农业发展的重要制约因素[1]. 另一方面,随着经济的发展,人口的增加和用水量的增大,导致污水排放量急剧上升,特别是未经过处理的废污水大量排放致使水污染加剧[2, 3]. 污水资源化利用是减轻污水排放二次污染和缓解农业用水紧张的发展趋势[4]. 我国农村人口众多,农村污水排放量大,污染程度低且能够最大限度实现回用. 但由于缺乏有效的处理设施,导致任意排放. 一方面,造成周边水体的富营养化,严重污染了农村的生态环境; 另一方面,农村污水直接用于农田灌溉,会造成作物品质和农田土壤性能降低,也成为疾病传染扩散的源头,容易导致水源性人畜共患疾病的发生与流行[5],直接威胁广大农民群众的身体健康并阻碍农村的经济发展. 鉴于水资源紧缺现象的不断加剧,为了确保农业生产,再生水应用已成为国内外缓解农业用水压力的重要措施之一[6]. 但是由于经济条件和处理技术原因,再生水中的污染物质去除并不彻底,主要包括氮磷元素和病原体等. 从再生水作为自然水体替代水源的角度出发,了解农村污水处理工艺过程中优势菌群和致病菌的分布情况,进而为改进处理技术和预防疾病的发生提供理论依据和保障措施.
目前农村污水的研究主要集中于污水处理技术的优化及污灌对土壤理化性质与作物品质的评价,针对整体工艺流程中微生物的多样性及相关病原菌的研究报道甚少. 基于16S rDNA基因的非培养技术为揭示自然环境中微生物种类和遗传多样性开辟了一条全新的途径,摆脱了传统的分离培养方法不能完整反映微生态系统全部信息的束缚[7, 8]. 因此本实验采用末端限制性片段长度多态(T-RFLP)和16S rDNA基因文库分析技术,对MBR工艺处理农村污水各阶段的细菌群落多样性进行研究,并通过荧光定量PCR方法对其中的弓形菌进行定量分析[9, 10],评价农村污水处理系统的有效性以及再生水资源化利用的可行性,以期为再生水灌溉的合理实施提供理论依据. 1 材料与方法 1.1 样品采集及处理
本研究中的污水处理厂位于北京市怀柔区桥梓镇北宅村,采用膜-生物反应器处理工艺(MBR,图 1),主要收纳处理当地居民的生活污水,日处理量600 m3. 整个处理工艺流程由原水集水井、调节池、好氧池、膜生物反应池和出水组成. 水样按照《生活饮用水标准检验方法水样的采集与保存》(GB/T 5750.2-2006)和《城镇污水处理厂污染物排放标准》(GB 18918-2002)每个处理阶段采集1~5 L. 采样容器材质为聚乙烯和硼硅玻璃,测定水质一般理化指标的采集容器经过10%硝酸或盐酸浸泡,取出用自来水反复冲洗,并用蒸馏水洗净放置烘箱烘干; 微生物指标采样容器用自来水冲洗数次,10%盐酸溶液浸泡过夜,依次用自来水和蒸馏水冲洗,之后容器经过高压蒸汽灭菌备用. 取样方式为深层水采样,利用上海雷磁DZB-718便携式多参数水质分析仪实时测定部分水质参数:温度、pH、溶解氧(DO)、电导率等,同时足量采集水样保存于4℃,24 h内送至谱尼公司(北京)测定其他水质参数和一些卫生指标(总大肠菌群和粪大肠菌群),检测方法参照国家标准,检测结果如表 1所示.
 | 图 1 膜-生物反应器处理工艺流程示意 Fig. 1 Process flow diagram of MBR |
| 表 1 MBR处理工艺流程水质特征1) Table 1 Water quality characteristics in MBR treatment process |
采集的水样保存于4℃冰盒,实验室于8 h内预处理. 原污水与再生水分别取100 mL和2 L用真空抽滤装置以0.22 μm微孔滤膜(Millipore,美国)过滤,其他水样各取5 mL低速离心收集沉淀,将滤膜与收集的沉淀立即放入-20℃冰箱保存用于微生物分析. 1.2 实验仪器
上海雷磁DZB-718便携式多参数水质分析仪; 湖南湘仪H165-W高速离心机; 北京东胜创新生物科技有限公司生产的东胜龙黑金刚EDC-810PCR仪; 美国安培公司生产的快速Fastprep-24核酸提取仪; 英国Syngene G: BOX 凝胶成像系统; 美国Thermo Fisher公司ND-2000核酸蛋白测定仪; 北京百晶生物技术有限公司BG-Power 600i电泳仪; 美国Agilent 公司生产的Strata gene Mx3005P 实时荧光定量PCR仪. 江苏太仓市科教器材厂HZ-9211KB恒温振荡器; 上海一恒科学仪器有限公司LRH-70生化培养箱; 郑州长城科工贸有限公司SHB-Ⅲ循环水式多用真空泵; 瑞士梅特勒-托利多国际股份有限公司电子天平AL104; 海门市其林贝尔仪器制造有限公司LX-200迷你离心机; 海门市其林贝尔仪器制造有限公司GL-88B漩涡混合器; 宁波新芝生物科技股份有限公司SB5200DT超声波清洗机. 1.3 基因组DNA提取
滤膜样品先用无菌剪刀剪碎,然后和其他样品一同用Fast DNA SPIN Kit for Soil试剂盒(MP Biomedicals,美国)提取总基因组DNA,具体步骤参照试剂盒说明书. 取5 μL基因组DNA 溶液用1%的琼脂糖凝胶电泳检测提取效果,采用NanoDrop2000(Thermo Scientific,美国)测定浓度,然后保存于-20℃冰箱备用. 1.4 16S rDNA基因克隆文库构建 1.4.1 基因组DNA的PCR扩增
以基因组DNA为模板,利用细菌16S rDNA基因的通用引物27F和1492R进行PCR扩增. 50 μL PCR反应体系:5 μL 10×PCR buffer (含20 mmol ·L-1 MgCl2),4 μL dNTP(10 mmol ·L-1),1 U Taq DNA聚合酶,1 μL DNA模板,灭菌的ddH2O补至50 μL.
PCR反应条件:95℃预变性10 min; 95℃变性45 s,55℃退火1 min,72℃延伸45 s,循环35次; 最后72℃延伸10 min,4℃保持. PCR扩增产物用1%的琼脂糖凝胶电泳检测. 1.4.2 16S rDNA基因克隆、转化和文库构建
将得到的PCR扩增产物用E.Z.N.ATM Gel Extraction Kit(OMEGA Biotek,美国) 进行纯化,回收后的DNA片段与pGEM-T Easy Vector(Promega,美国)在4℃条件下连接16 h. 连接产物导入感受态大肠杆菌DH5α,加入液体LB培养基于37℃ 200 r ·min-1培养3 h,取100 μL涂布于LB(含Amp/IPTG/X-Gal)平板上,37℃条件下培养18 h后进行蓝白斑筛选. 用无菌牙签随机挑取白色单菌落进行转板培养,以载体引物SP6/T7进行PCR扩增. PCR扩增产物用1%琼脂糖凝胶电泳检测,筛选阳性克隆子,构建16S rDNA基因克隆文库. 原污水中细菌数量较多,随机挑取118个阳性克隆子进行PCR,PCR产物用DNA限制性内切酶HhaI(Promega,美国)按照说明书条件酶切分型,酶切产物用2%的琼脂糖凝胶进行电泳,电压60 V,时间2 h. 电泳结束后EB染色,再用蒸馏水漂洗. 用Stratagene G:BOX 凝胶成像系统检测并保存结果. 图像用Quantity ONE 4.3.0软件进行分析. 确定ARDRA类型数目,每个ARDRA分型结果作为一个OTU,从每个OTU中选取1个克隆送北京睿博兴科生物技术有限公司进行测序. 1.4.3 16S rDNA基因克隆文库多样性分析
测序结果在 Bellerophon网站分析可能存在的嵌合体并去除,在核酸相似性≥97%水平上划分操作分类单元(operational taxonomic unit,OTU). 基因文库的多样性覆盖率(coverage)通过公式c=1-n1/N[11]计算,式中,N代表基因文库的克隆数,n1为仅有一个克隆的OTU数目. 百分率越高表明文库多样性覆盖率越高. 对基因文库的序列做多样性指数分析,Shannon-Wiener 指数(H)和Simpson指数(D)计算公式为[12, 13]:
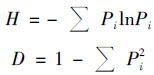
通过将测序成功的阳性克隆序列与NCBI数据库进行Blast同源性检索后,下载同源性最高的序列和部分已知种属的同源性最高的序列作为参考,使用Mega 5.0软件包,以邻接法(Neighbor Joining Analysis)进行系统发育树的构建. 发育树的稳定性Bootstraps来分析评估,树的节点通过非参数支持率计算(1 000次重复运算). 1.5 T-RFLP分析
利用具有荧光标记的细菌通用引物27F/926R(引物27F的5′端用FAM标记)扩增水样基因组DNA. PCR反应条件:95℃预变性5 min; 95℃变性45 s,50℃退火45 s,72℃延伸1 min,30个循环; 最后72℃延伸10 min,4℃保持. 两次PCR产物进行混合,以减少扩增带来的误差. PCR产物用E.Z.N.ATM Gel Extraction Kit试剂盒进行纯化.
纯化后的PCR产物用RsaI(Takara,Japan)进行酶切. 10 μL酶切体系:RE 10×buffer 1 μL,Acetylated BSA(10 μg ·μL-1)0.1 μL,DNA~100 ng,混匀后加入RsaI(10 U ·μL-1)0.25 μL,然后加ddH2O补齐至10 μL. 酶切条件:37℃酶切3 h.
T-RFLP图谱用GeneMarker(Version 1.71)软件进行分析,选择的内标为GS1200liz. 对于细菌,由于相对数量过小的限制性末端片段不会对群落的特性产生明显的影响[9, 10, 14],故在分析中舍去了相对数量<1%的T-RFs. 根据T-RFLP图谱计算微生物群落结构多样性Shannon香农指数(H)、Simpson辛普森指数(D)、物种均度(E)及丰度(S). T-RFLP图谱上的峰面积大小和数量反映了微生物群落结构和多样性,每个峰可以作为一个OTU来进行分析. 1.6 实时荧光定量PCR
利用病原菌特异性引物对水样基因组DNA进行常规PCR扩增,纯化回收PCR产物. 产物与pGEM-T Easy载体连接后导入感受态细胞DH5α,涂板筛选. 经PCR鉴定为阳性的菌落转入LB液体培养基扩大培养,用E.Z.N.A.®Plasmid Mini Kit I Spin Kit (Omega Bio-tek)试剂盒提取质粒. 利用NanoDrop2000(Thermo Scientific,美国) 测定质粒浓度,将已知拷贝数的质粒10倍梯度稀释作为标准模板,利用GoTaq® qPCR Master Mix with SYBR Green® dye(Promega,美国)对上述菌的目的基因进行定量检测,所用引物如表 2所示.
| 表 2 实验所用引物及基因片段大小 Table 2 Primers used in the experiment and amplicon size |
反应体系:12.5 μL Go Taq qPCR Master Mix(Promega,美国),上下游引物各0.5 μL,模板1 μL,加无菌ddH2O补齐至25 μL,反应于八联排管(Axygen,美国)中进行. 荧光定量PCR程序为:95℃ 2 min; 94℃ 15 s,60℃ 30 s,72℃ 45 s,40个循环; 72℃ 10 min. 熔解曲线条件:95℃ 1 min,55℃ 30 s,95℃ 30 s,所有反应均设置3个重复. 2 结果与讨论 2.1 污水处理工艺水质指标及处理效果分析
MBR处理工艺各阶段水质参数如表 1所示. 进水与出水相比,COD、总氮、氨氮及总磷去除率分别达到91.7%、20.9%、97.5%和21.9%. 各处理阶段中亚硝态氮均未检出,硝态氮、溶解氧、电导率有些波动但相对稳定. 出水中的指示菌总大肠菌群和蛔虫卵均未检出,粪大肠菌群≤3个 ·L-1. 出水各项指标均达到《城镇污水处理厂污染物排放标准》(GB 18918-2002)一级标准和《农田灌溉水质标准》(GB 5084-2005). 2.2 16S rDNA基因克隆文库建立
实验共挑取118个阳性克隆,酶切分型后分为14个OTU用于测序,其中10个OTU测序成功,即有73个阳性克隆测序成功.
16S rDNA基因克隆文库细菌Shannon-Wiener多样性指数为1.85,物种均匀度0.80,表明未处理的农村污水中某类优势菌种存在比例较高. 本研究中N=73,n1=1,克隆文库的覆盖度为0.986,表明克隆文库中包含的微生物种类占样品中全部微生物的种类比例已相当高,能较全面反映原污水中细菌群落多样性.
10个OTU序列的BLAST比对结果见表 3,下载同源性最高的序列作为参考. OTU序列与GenBank数据库中已知细菌的16S rDNA基因序列相似性最高为99%,最低为93%; 其中有8个OTU(80%)相似性高于97%,2个OTU(20%)相似性低于94%. 在序列同源性研究中,一般认为细菌16S rDNA基因序列同源性低于97%即属于不同的种.
| 表 3 GenBank中与克隆子序列最相似菌株 Table 3 Similar strains with the subsequence of cloning in GenBank |
将农村污水中细菌16S rDNA基因文库菌株进行系统分类,可分为3门、4纲、4目、4科、6属. 在73个克隆中,有67个克隆子属于变形菌门、2个属于厚壁菌门、1个属于拟杆菌门,还有3个为未培养细菌. 变形菌门占总数的91.8%,是克隆文库中的第一大类群. 其中ε-变形菌纲为71.2%,β-变形菌纲为20.5%. 该样品中优势菌属是ε-变形菌纲的弓形菌属,占克隆总数的68.5%,与吴春笃等[19]、王峰等[20]的研究结果相符. 拟杆菌门(Bacteroidetes)、厚壁菌门(Firmicutes)和放线菌门(Actinobacteria)等类别的细菌一般比例也在5%~20%[21, 22, 23, 24]. 本文的研究对象农村污水中含有大量的厕所粪便黑水,弓形菌作为导致肠道疾病的病原菌生长适应性强,目前已知的3种致病性弓形菌分别分离自粪便及血液中. 由此推断农村污水的来源及成分营造的生境利于共性的生长繁殖导致数量较高. 除了变形菌门,厚壁菌门占2.70%,拟杆菌门占1.40%,未培养细菌为4.10%. 表明所研究的农村污水内的细菌有极丰富的生理多样性,并潜藏着特有的微生物资源,有待于进一步深入研究.
由OTU代表克隆序列和BLAST中相似菌株的16S rDNA基因序列绘制的系统进化树见图 2.
 | 图 2 农村污水微生物系统进化树 Fig. 2 Phylogenetic tree of microorganisms in rural sewage |
由图 2可知,10个OTU分别与属于Proteobacterium类群、Firmicutes类群、Bacteroidetes类群的已知细菌聚类在一起. 测得的未知类群OTU9聚类在拟杆菌门,与Bacteroides fragilis相距较近. 2.3 T-RFLP结果分析
MBR处理工艺各阶段细菌群落的T-RFLP图谱如图 3所示(取DNA片段长度处于50~1 000 bp,并且荧光强度>50RFU的峰). 通过对不同处理阶段的图谱比较分析,可以看出调节池、好氧池和膜生物反应池3个系统中的T-RFs片段数量较多,并且群落结构在一定程度上保持相对稳定,反映该处理系统本身具有一定的缓冲能力.
 | 图 3 不同处理阶段农村污水细菌群落T-RFLP 图谱 Fig. 3 T-RFLP maps in different treatment stages of rural wastewater |
利用目前公认的数据库MiCA(http://mica.ibest.uidaho.edu/)在线比对,得出部分T-RFs片段代表的类群及其在MBR不同处理过程中的存在情况,如表 4 所示. 结合T-RFLP图谱和表 4可以看出,调节池中微生物种类最多,然后是出水、好氧池、膜池、进水,与T-RFLP图谱有一定差别,这是因为部分不同物种片段大小重叠无法确定其类别. 出水中虽然微生物种类较多,但结合T-RFLP图谱看,可知其所占比例较小. 对已确定的菌株,T-RF(89、90、98、209、211、585 bp)仅存在于进水中,说明其在后续处理过程中被除去. 在进水中所占比例较高的T-RF-95bp鉴定为Terriglobus sp.. T-RF-205 bp在各阶段均存在且在调节池、好氧池中所占比例较高,经鉴定为Clostridium sp.. 另外一个在调节池、好氧池中占较高比例的T-RF-55 bp鉴定为Streptococcus sp.. 膜池中占较高比例的T-RF-57 bp、T-RF-605 bp分别为Mitsuokella sp.和Kineococcus sp.,而T-RF-358 bp和T-RF-765 bp未能鉴定出物种类型. 出水中占较高比例的T-RF-605 bp和T-RF-765 bp都未能鉴定出物种类型.
| 表 4 部分T-RF所代表的菌株1) Table 4 Strains represented by T-RFs |
根据T-RFLP数据计算得到的多样性指数如图 4所示. 不同阶段水体细菌的多样性指数不同,其中调节池的Shannon-Wiener多样性指数、物种丰度、均匀度最高,分别为3.56、43.0和0.95,表明调节池系统内的细菌种类多样性最高且物种分配最均匀,原因是因为调节池作为整个处理流程的前段工艺,主要作用是调节水量和水质,为微生物生长提供了繁殖条件,能够在污水处理过程中起到良好的调节缓冲作用; 好氧池和膜池在取样期间有轻微的污泥膨胀现象,污泥活性的高低可能对不利用某些类群微生物的生长. 所有指数在调节池阶段之后均随处理流程呈现出递减趋势,出水系统的多样性指数及物种丰度低于其他处理阶段,表明MBR处理工艺对农村污水中微生物的去除效果较好.
 | 图 4 不同处理阶段污水多样性指数 Fig. 4 Diversity indices in different treatment stages of rural wastewater |
根据16S rDNA基因克隆文库结果,原污水中弓形菌为优势菌属,因此对MBR处理系统中的弓形菌属和细菌总数进行荧光定量分析. 弓形菌属和总细菌的扩增产物片段大小分别为331 bp、315 bp,凝胶图谱如图 5所示. 定量结果显示,扩增效率接近100%,相关系数(R2)都在99%以上,符合定量要求. 根据标准曲线计算MBR处理系统进水与出水中细菌的基因拷贝数如表 5所示.
 | 1.空白对照;2.弓形菌属;3.细菌总数 图 5 病原菌扩增产物凝胶图谱 Fig. 5 Gel maps of pathogen amplification product |
| 表 5 样品中病原菌基因拷贝数/copies ·L-1 Table 5 Copy numbers of pathogenic genes in the samples/copies ·L-1 |
微生物群落结构对MBR处理系统的稳定运行和有效去除理化污染物起着重要作用[25],但定量结果显示出水中含有大量病原菌,说明MBR工艺对弓形菌去除效果不太理想,虽然膜组件可以拦截大部分细菌、病毒,如果未经消毒处理,出水中仍有大量病原菌残留.
弓形菌是近几十年发现的人畜共患致病菌,大小为0.2~0.9 μm×0.5~3 μm,革兰氏阴性,无芽孢,无荚膜[26]. 弓形菌为变形菌门(Proteobacteria)ε-变形菌纲(ε-Proteobacteria)弯曲杆菌目(Campylobacterales)弯曲杆菌科(Campylobacteraceae)的一个新属,其中有3个种具有致病性,分别为布氏弓形菌(A. butzleri)、嗜低温弓形菌(A. cryaerophilus)、斯氏弓形菌(A. skirrowii). 16S rDNA基因克隆文库结果显示,弯曲杆菌科中共有50个弓形菌属,其中有40个为嗜低温弓形菌,1个为斯氏弓形菌,为农村污水中第一优势种群. 在过去几十年里,弓形菌在污水、地表水、地下水及畜禽肉类产品中均有发现,腹泻病人的粪便中[27, 28, 29]、菌血症病人[28]、肝硬化病人[30]和急性坏疽性阑尾炎病人[31]的血液中等都发现有弓形菌的踪迹. 但目前国内外对弓形菌致病机制和潜在毒性因子鲜有报道,这可能是与弓形菌没有如沙门氏菌和弯曲杆菌列入临床常规检测项目有关[32]. 3 结论
(1)通过对污水处理厂不同处理阶段水体微生物群落T-RFLP图谱的直观分析可知,农村污水中的微生物群落结构较为复杂,并且在一定程度上群落保持相对稳定. 其中调节池污水中微生物多样性和物种分配均匀度最高.
(2)对污水处理厂进水的克隆文库分析表明本研究农村污水内细菌具有较高的多样性,并潜藏着特有的微生物资源. 变形菌门(Proteobacteria)是污水处理系统中数量最多、种类最丰富的细菌类群,在相关研究中其比例都在50%以上. 结合定量PCR结果可知,农村污水中存在着大量细菌,致病菌-弓形菌的比例较高. MBR处理系统的所有出水物化指标均接近一级A排放标准,病原指示菌的数量虽然明显减少,但弓形菌的去除效果还不够理想,其数量仍为108copies ·L-1. 可见,MBR处理后的污水直接用于灌溉,也可能会对人体健康造成潜在危害. 因此,污水资源化安全利用需要污水处理技术和适宜的灌溉方式有机结合,从而降低再生水灌溉后病原菌所带来的健康风险,改善农村生态环境并保障粮食安全. 未经消毒处理的再生水灌溉后,病原菌在作物及土壤中的迁移转化及残留还需要进一步验证.
| [1] | 宰松梅, 王朝辉, 庞鸿宾. 污水灌溉的现状与展望[J]. 土壤, 2006, 38 (6): 805-813. |
| [2] | 张建云, 贺瑞敏, 齐晶, 等. 关于中国北方水资源问题的再认识[J]. 水科学进展, 2013, 24 (3): 303-310. |
| [3] | 刘喜峰, 徐红松, 宋文娟. 我国北方地区水资源问题及对策[J]. 河南水利与南水北调, 2007, (5): 21-22. |
| [4] | 焦志华, 黄占斌, 李勇, 等. 再生水灌溉对土壤性能和土壤微生物的影响研究[J]. 农业环境科学学报, 2010, 29 (2): 319-323. |
| [5] | 邹翔, 刘菊萍, 张小生. 规模化养殖场面源污染问题调查及治理对策研究[J]. 南方农机, 2009, (5): 36, 47. |
| [6] | 周新伟, 沈明星, 陆长婴, 等. 再生水灌溉对白菜产量、品质及土壤化学性状的影响[J]. 江西农业科学, 2014, 42 (1): 130-133. |
| [7] | 徐成斌, 孟雪莲, 马溪平, 等. 16S rDNA克隆文库方法对制药废水处理系统中微生物多样性的研究[J]. 生态环境学报, 2009, 18 (4): 1236-1240. |
| [8] | 董萍, 孙寓姣, 王红旗, 等. 利用T-RFLP技术对温榆河微生物群落结构研究[J]. 中国环境科学, 2011, 31 (4): 631-636. |
| [9] | 史青, 柏耀辉, 李宗逊, 等. 应用T-RFLP技术分析滇池污染水体的细菌群落[J]. 环境科学, 2011, 32 (6): 1786-1792. |
| [10] | 滕齐辉, 曹慧, 崔中利, 等. 太湖地区典型菜地土壤微生物16S rDNA的PCR-RFLP分析[J]. 生物多样性, 2006, 14 (4): 345-351. |
| [11] | Good I J. The population frequencies of species and the estimation of population parameters[J]. Biometrika, 1953, 40 (3-4): 237-264. |
| [12] | Elshahed M S, Senko J M, Najar F Z, et al. Bacterial diversity and sulfur cycling in a mesophilic sulfide-rich spring[J]. Applied and Environmental Microbiology, 2003, 69 (9): 5609-5621. |
| [13] | 余素林, 吴晓磊, 钱易. 环境微生物群落分析的T-RFLP技术及其优化措施[J]. 应用与环境生物学报, 2006, 12 (6): 861-868. |
| [14] | 王洪媛, 管华诗, 江晓路. 微生物生态学中分子生物学方法及T-RFLP技术研究[J]. 中国生物工程杂志, 2004, 24 (8): 42-47. |
| [15] | Galkiewicz J P, Kellogg C A. Cross-kingdom amplification using Bacteria-specific primers: complications for studies of coral microbial ecology[J]. Applied and Environmental Microbiology, 2008, 74 (24): 7828-7831. |
| [16] | Muyzer G, Teske A, Wirsen C O, et al. Phylogenetic relationships of Thiomicrospira species and their identification in deep-sea hydrothermal vent samples by denaturing gradient gel electrophoresis of 16S rDNA fragments[J]. Archives of Microbiology, 1995, 164 (3): 165-172. |
| [17] | Kingombe C I B, Huys G, Tonolla M, et al. PCR detection, characterization, and distribution of virulence genes in Aeromonas spp.[J]. Applied and Environmental Microbiology, 1999, 65 (12): 5293-5302. |
| [18] | Castillo M, Martín-Orúe S M, Manzanilla E G, et al. Quantification of total bacteria, enterobacteria and lactobacilli populations in pig digesta by real-time PCR[J]. Veterinary Microbiology, 2006, 114 (1-2): 165-170. |
| [19] | 吴春笃, 许小红, 宁德刚, 等. 城市污水细菌多样性及其生物安全性研究[J]. 中国安全科学学报, 2008, 18 (1): 119-122. |
| [20] | 王峰, 傅以钢, 夏四清, 等. PCR-DGGE技术在城市污水化学生物絮凝处理中的特点[J]. 环境科学, 2004, 25 (6): 74-79. |
| [21] | Chouari R, Le Paslier D, Daegelen P, et al. Molecular analyses of the microbial community composition of an anoxic basin of a municipal wastewater treatment plant reveal a novel lineage of proteobacteria[J]. Microbial Ecology, 2010, 60 (2): 272-281. |
| [22] | Del Casale A, Flanagan P V, Larkin M J, et al. Analysis of transduction in wastewater bacterial populations by targeting the phage-derived 16S rRNA gene sequences[J]. FEMS Microbial Ecology, 2011, 76 (1): 100-108. |
| [23] | Kwon S, Kim T S, Yu G H, et al. Bacterial community composition and diversity of a full-scale integrated fixed-film activated sludge system as investigated by pyrosequencing[J]. Journal of Microbiology and Biotechnology, 2010, 20 (12): 1717-1723. |
| [24] | 宋洪宁, 杜秉海, 张明岩, 等. 环境因素对东平湖沉积物细菌群落结构的影响[J]. 微生物学报, 2010, 50 (8): 1065-1071. |
| [25] | Hallin S, Lydmark P, Kokalj S, et al. Community survey of ammonia-oxidizing bacteria in full-scale activated sludge processes with different solids retention time[J]. Journal of Applied Microbiology, 2005, 99 (3): 629-640. |
| [26] | 毕水莲, 孟赫诚. 致病性弓形杆菌属生物学特性及诊断研究进展[J]. 现代食品科技, 2013, 29 (1): 211-214. |
| [27] | Lerner J, Brumberger V, Preac-Mursic V. Severe diarrhea associated with Arcobacter butzleri[J]. European Journal of Clinical Microbiology & Infectious Diseases, 1994, 13 (8): 660-662. |
| [28] | Hsueh P R, Teng L J, Yang P C, et al. Bacteremia caused by Arcobacter cryaerophilus 1B[J]. Journal of Clinical Microbiology, 1997, 35 (2): 489-491. |
| [29] | Kiehlbauch J A, Brenner D J, Nicholson M A, et al. Campylobacter butzleri sp. nov. isolated from humans and animals with diarrheal illness[J]. Journal of Clinical Microbiology, 1991, 29 (2): 376-385. |
| [30] | Yan J J, Ko W C, Huang A H, et al. Arcobacter butzleri bacteremia in a patient with liver cirrhosis[J]. Journal of the Formosan Medical Association, 2000, 99 (2): 166-169. |
| [31] | Lau S K P, Woo P C Y, Teng J L L, et al. Identification by 16S ribosomal RNA gene sequencing of Arcobacter butzleri bacteraemia in a patient with acute gangrenous appendicitis[J]. Molecular Pathology, 2002, 55 (3): 182-185. |
| [32] | Lehner A, Tasara T, Stephan R. Relevant aspects of Arcobacter spp. as potential foodborne pathogen[J]. International Journal of Food Microbiology, 2005, 102 (2): 127-135. |