 2015, Vol. 36
2015, Vol. 36


2. 青海黄河上游水电开发有限公司西宁发电分公司, 西宁 810007
2. Qinghai Huanghe Hydropower Development Co., Ltd., Xining 810007, China
整体煤气化联合循环发电(IGCC)技术把高效的燃气-蒸汽联合循环发电系统与洁净的煤气化技术结合起来,既有高发电效率,又有极好的环保性能,是一种有发展前景的洁净煤发电技术[1]. 在目前技术水平下,IGCC发电的净效率可达43%~45%,而污染物的排放量仅为常规燃煤电站的1/10,环境效益明显[2]. 煤气化过程中仍有大量有害的汞释放出来,并且主要以单质汞形态存在,煤气化衍生物气体中汞浓度也要大于燃煤电厂尾气中汞浓度[3].
目前对燃煤烟气下的单质汞脱除的吸附剂或催化剂主要有飞灰、 活性炭以及改性活性炭等碳基吸附剂,矿石类吸附剂和金属及金属氧化物[4,5]. 它们存在活性温度窗口较窄,在中高温(300~450℃)下脱汞效率较低的缺点[4,6],无法用于IGCC煤气化中的汞的脱除.
分子筛具有高硅铝比和特殊孔道结构,因为其独特的孔道结构和酸性成为重要的催化材料. 为了得到在中高温(300~450℃)下单质汞脱除效率较高的催化剂,本研究用分子筛作为载体,采用浸渍法在分子筛上负载活性锰、 铈组分得到Mn-Ce/分子筛复合催化剂,对催化剂的物理化学特性进行了表征,分析了该催化剂在300~450℃下氧化单质汞的性能及机制,以期为中高温下可以高效氧化单质汞的催化剂提供参考. 1 材料与方法 1.1 催化剂的制备与表征
本实验采用硅铝比(Si/Al)为50的分子筛作为载体(M50),以过渡金属锰和铈的氧化物作为催化剂的活性组分,采用浸渍法进行负载改性制备实验所需的 Mn-Ce/M50催化剂. 首先将一定量的M50分子筛置于马弗炉中于550℃高温焙烧4 h,以去除分子筛制备过程中内部残留的有机模板剂及杂质,并进行活化. 焙烧完毕后于马弗炉中自然冷却,温度降至100~150℃后将分子筛转移至干燥器中,得到实验所用的催化剂载体.
实验中催化剂的负载量的多少是根据所用物质溶解形成一定浓度的溶液换算,实验所需的催化剂的负载量指理论上活性组分占催化剂总质量的质量分数,由于MnOx的O量不确定,因此该催化剂的负载量以锰计算. 样品标记为(y,a) Mn-Ce/M50,其中y表示负载的Ce和Mn的质量比,设需制备Mn负载量为a%的催化剂. 具体的制备步骤如下:①用分析天平称取定量的四水氯化锰、 六水硝酸铈置于烧杯中,采用移液管量取一定量去离子水倒入烧杯中,搅拌使上述试剂完全溶解作为浸渍分子筛的溶液. ②用分析天平称量一定量的分子筛,加入上述溶液中,使得分子筛完全浸渍,并机械搅拌60 min. ③将上述样品置于洁净的鼓风干燥箱内于60℃条件下干燥24 h. ④将以上干燥好的氯化锰、 硝酸铈浸渍的M50分子筛分为3份,在空气气氛下于马弗炉中分别于350、 450、 550℃焙烧4 h. ⑤研磨,过80目筛储存备用.
X-射线光电子能谱(XPS)采用VG Multilab 2000型能谱仪,扫描模式为FRR,测试条件为MgKα靶. XPS测试前需将催化剂在110℃下干燥,结果根据C1s 结合能 (BE) 为284.6 eV标定,最终得到催化剂的XPS光谱图. 1.2 实验装置与方法
在小型脱汞实验台(图 1)进行脱汞实验,模拟烟气用SO2、 NO、 N2、 O2、 CO2等气瓶进行配制,汞蒸气发生装置主要由恒温槽、 汞渗透管、 U型石英管组成. 各组分气体分别用流量计进行定量,气体总流量为2.0 L ·min-1,O2、 CO2的体积浓度分别为6%和12%,汞浓度为35.0 μg ·m-3,每次实验的吸附剂用量为1.0g. 反应器放在可程序控制升温的加热炉内,通过设定加热炉的温度来控制反应温度. 通过反应器反应后的模拟烟气由俄罗斯Lumex公司的RA-915M测汞仪进行检测,该仪器可实时连续监测单质汞的浓度,时间分辨率为1 s.
采用公式(1)来计算反应器中单质汞的氧化效率Eoxi:
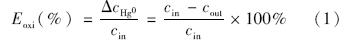
 | 图 1 实验系统示意
Fig. 1 Experimental setup
|
Mn-Ce/M50催化剂中Mn2p和Ce3d的XPS光谱如图 2所示. 图 2(a)中Mn2p 3/2和Mn 2p 1/2的特征峰的结合能范围为630~660 eV. Mn2p 3/2的特征峰含两个次峰,一个是Mn4+的特征(642.7 eV左右),一个是Mn3+的特征峰(641.2 eV左右)[7]. 由于Mn4+比Mn3+的氧化性更强,所以Mn4+/Mn3+比值越高越利于单质汞的氧化. 如图 2(b)所示,标示为ν的峰对应于Ce3d 5/2状态[8,9],其中ν和ν2 峰对应于3d104f0电子状态的Ce4+,而ν1峰对代表处于3d104f1 电子状态的Ce3+[10, 11]. 这表明Mn-Ce/M50催化剂表面存在Ce4+与Ce3+,Ce3+的存在造成了催化剂表面的电荷不平衡,形成了电子空穴及一些游离的化学键[10,11],因而使得催化剂表面富集了大量的化学吸附态氧. Mn-Ce/M50催化剂表面的Mn4+形成了较多的晶格氧以及Ce3+产生的化学吸附态氧在氧化过程中具有很高的活性[12].
 | 图 2 XPS光谱图
Fig. 2 XPS spectra
|
为了解焙烧温度对制备的(0.5,6)Mn-Ce/M50催化剂氧化烟气中单质汞的影响,本实验在催化剂制备过程中,样品浸渍干燥后分别采用350、 450、 550℃焙烧制备了3种同一负载量的催化剂. 实验过程中采用3个不同活化温度制备的催化剂进行了脱汞实验,催化剂用量1 g. 考察其在一定的反应温度范围内的脱汞规律及效果.
由图 3可知,350℃的反应温度下3种焙烧温度对(0.5,6)Mn-Ce/M50催化剂的氧化单质汞的性能影响较大. 在450℃和550℃这两种焙烧温度下制得的催化剂的脱汞效果比350℃的焙烧温度下制得的催化剂效果好. 350℃的焙烧温度条件下制得的催化剂氧化汞的性能较低,这是由于焙烧温度较低时负载组分不能完全分解转变为活性组分; 而且负载组分会占据分子筛表面,降低分子筛载体的吸附性能. 焙烧温度升高到450℃和550℃后,分子筛表面浸渍的负载物可以完全分解成对单质汞氧化性较好的金属氧化物. 但是催化剂的焙烧温度过高会破坏分子筛的结构,使分子筛的酸性位减少,从而降低了吸附性能; 同时高温焙烧的MnCl2受热分解,所形成的锰氧化物中的锰离子会转变成较低价态的锰,对单质汞的氧化作用减弱[13, 14, 15]. 因此,在催化剂焙烧时550℃的焙烧温度比较合适,后面的催化剂主要是在550℃的焙烧温度下制得的.
 | 实验1 h对应的效率,下同 图 3 不同活化温度下的脱汞效率 Fig. 3 Oxidation efficiency of Hg0 at different activation temperature |
为了摸清所制备的催化剂对单质汞的活性温度范围,本实验在300~450℃的反应温度范围内对(0.5,6)Mn-Ce/M50(焙烧温度为550℃)进行了脱汞实验,催化剂用量1.0 g,结果见图 4.
 | 图 4 反应温度对Hg0去除的影响
Fig. 4 Effect of reaction temperature on the oxidation efficiency of Hg0
|
从图 4可以发现在300~450℃的反应温度范围内,(0.5,6)Mn-Ce/M50随温度升高单质汞的氧化效率呈下降的趋势,在300℃的反应温度下催化剂的脱汞效率最高,可以达到93%. 300~450℃温度条件下其脱汞性能比较稳定,保持在80%以上,在450℃时催化剂的脱汞效率最低(为80%). 这表明催化剂对汞的氧化过程符合Langmuir-Hinshelwood机制[16],单质汞先被吸附到催化剂表面,然后吸附态的单质汞再与活性物质发生反应. Langmuir-Hinshelwood机制可以解释 Mn-Ce/M50催化剂对汞的氧化效率随温度升高而降低,主要是由于催化剂对单质汞的吸附能力降低会导致汞的氧化效率也随之降低,即使催化剂本身没有变化.
为了便于进一步机制分析,对焙烧温度为550℃下制得的(6)Mn/M50、 (3)Ce/M50、 (0.5,6)Mn-Ce/M50的脱汞特性进行了研究,结果见图 5.
 | 图 5 (0.5,6)Mn-Ce/M50、 (6)Mn/M50、 (3)Ce/M50、 M50氧化单质汞的效率
Fig. 5 Oxidation efficiency of Hg0 by different catalysts
|
从图 5可以发现M50基本没有脱汞效果,而Mn/M50与Ce/M50催化剂对单质汞的氧化效率都低于Mn-Ce/M50的氧化效率. Mn与Ce负载到分子筛载体上得到了更高的单质汞的氧化效率,Mn与Ce的同时负载产生了协同促进作用,这表明分子筛中引入活性组分锰和铈后,分子筛的脱汞性能得到很大提高.
有研究发现负载在吸附剂上的MnCl2经过焙烧后的主要活性成分为锰氧化物(MnOx)[17],根据实验结果和Mars-Maessen机制[18],推断MnOx在单质汞的氧化过程中起到重要的作用[公式(2)~(6)],因为单质汞的氧化过程会有电子转移,气态的 Hg0 在催化剂表面被物理吸附生成Hg0(ad),吸附态的单质汞可以与金属氧化物 (MxOy)的晶格氧发生反应生成HgO,消耗的晶格氧可以由气态氧补给.

CeO2 由于具有强大的储氧能力及可以在不同气氛条件下实现Ce3+/Ce4+转换[12,19],(0.5,6)Mn-Ce/M50催化剂中CeO2可以起到辅助剂的作用,既可以提供氧空位,可以拓宽催化剂的活性温度范围. 结合前人研究成果[8,20]和本研究的XPS分析结果,发现MnOx与CeO2的结合促进了活性物质在Mn-Ce/M50表面的分布,同时产生了更多的活性氧,正是这些活性物质促进了单质汞的氧化. 2.4 SO2的影响
不同的文献对SO2对单质汞氧化的影响报道不同,有的发现起促进作用[3],有的发现起抑制作用[21]. 如图 6所示,在CO2+O2+N2载气中添加3000 mg ·m-3的SO2后,Eoxi从92%降低到72%,这说明SO2抑制了Mn-Ce/M50上单质汞的氧化. 这可能是由于以下3个原因引起的:①催化剂表面单质汞的氧化是通过Langmuir-Hinshelwood机制进行,只有吸附态的Hg0 才能被氧化,SO2与Hg0在催化剂表面发生竞争吸附,抑制了Hg0的吸附,从而抑制Hg0的氧化; ②SO2 与催化剂表面的活性氧反应生成了SO3,从而消耗了本可以氧化单质汞的活性氧; ③SO2与Mn-Ce/M50发生反应生成硫酸锰[22,23]和硫酸铈[24],导致催化剂中毒.
 | 图 6 SO2对Hg0氧化脱除的影响
Fig. 6 Effect of SO2 on the oxidation removal efficiency of Hg0
|
如图 7所示,在CO2+O2+N2载气中添加200mg ·m-3的NO后,单质汞的氧化率从92%降低到76%,这表明NO对单质汞的氧化有一定的抑制作用. 研究发现NO在催化剂表面可以被氧化成NO+及NO2等物质[25],由于NO消耗烟气中参与汞氧化的活性物质而抑制汞氧化. 也有研究发现NO可以弱吸附于MnOx-CeO2催化剂上,并与MnOx-CeO2催化剂反应生成少量的NO2、 亚硝酸根、 硝酸根等物质[26]. 实验中模拟烟气中NO的浓度为200mg ·m-3,比模拟烟气中汞的浓度高很多,
 | 图 7 NO对Hg0氧化脱除的影响
Fig. 7 Effect of NO on the oxidation removal efficiency of Hg0
|
3 结论
(1)分子筛中引入活性组分锰和铈后,分子筛氧化单质汞的性能得到很大提高,而且MnOx与CeO2混合后对单质汞的氧化产生了协同促进作用.
(2)Mn-Ce/M50上单质汞的催化氧化符合Langmuir-Hinshelwood机制. XPS结果表明Mn-Ce/M50催化剂表面存在 Mn4+、 Mn3+、 Ce4+和Ce3+,它们会促进单质汞的氧化.
(3)300~450℃的反应温度内,随温度升高单质汞的氧化效率呈下降趋势,在450℃时催化剂的脱汞效率最低. SO2及NO降低了单质汞的氧化效率,SO2及NO与Hg0在催化剂表面发生竞争吸附,抑制了Hg0的吸附,从而抑制Hg0的氧化; SO2及NO消耗本可以氧化单质汞的活性氧从而抑制单质汞的氧化.
| [1] | 施强, 乌晓江, 徐雪元, 等. 整体煤气化联合循环(IGCC)发电技术与节能减排[J]. 节能技术, 2009, 27 (1): 18-20. |
| [2] | Pavlish J H, Hamre L L, Zhuang Y. Mercury control technologies for coal combustion and gasification systems [J]. Fuel, 2010, 89 (4): 838-847. |
| [3] | Liu Y, Bisson T M, Yang H Q, et al. Recent developments in novel sorbents for flue gas clean up [J]. Fuel Processing Technology, 2010, 91 (10): 1175-1197. |
| [4] | 谭增强, 牛国平. 烟气汞脱除的研究进展[J]. 热力发电, 2013, 42 (10): 1-8. |
| [5] | Lee W, Bae G N. Removal of Elemental Mercury (Hg(0)) by Nanosized V2O5/TiO2 Catalysts[J]. Environmental Science & Technology, 2009, 43 (5): 1522-1527. |
| [6] | Wendt J O L, Lee S J. High-temperature sorbents for Hg, Cd, Pb, and other trace metals: Mechanisms and Applications [J]. Fuel, 2010, 89 (4): 894-903. |
| [7] | Qiao S H, Chen J, Li J F, et al. Adsorption and catalytic oxidation of gaseous elemental mercury in flue gas over MnOx/Alumina [J]. Industrial & Engineering Chemistry Research, 2009, 48 (7): 3317-3322. |
| [8] | Li H L, Wu C Y, Li Y, et al. Superior activity of MnOx-CeO2/TiO2 catalyst for catalytic oxidation of elemental mercury at low flue gas temperatures[J]. Applied Catalysis B: Environmental, 2012, 111-112 : 381-388. |
| [9] | Watanabe S, Ma X L, Song C S. Characterization of Structural and Surface Properties of Nanocrystalline TiO2-CeO2 Mixed Oxides by XRD, XPS, TPR and TPD[J]. The Journal of Physical Chemistry C, 2009, 113 (32): 14249-14257. |
| [10] | Gao X, Jiang Y, Fu Y C, et al. Preparation and characterization of CeO2/TiO2 catalysts for selective catalytic reduction of NO with NH3[J]. Catalysis Communications, 2010, 11 (5): 465-469. |
| [11] | Gao X, Jiang Y, Zhong Y, et al. The activity and characterization of CeO2-TiO2 catalysts prepared by the sol-gel method for selective catalytic reduction of NO with NH3[J]. Journal of Hazardous Materials, 2010, 174 (1-3): 734-739. |
| [12] | Li H L, Wu C Y, Li Y, et al. CeO2-TiO2 Catalysts for Catalytic Oxidation of Elemental Mercury in Low-Rank Coal Combustion Flue Gas[J]. Environmental Science & Technology, 2011, 45 (17): 7394-7400. |
| [13] | Mei Z J. The study of Co、Mn series sorbents for the removal of element mercury from simulated flue gas [D]. Shanghai: Shanghai Jiao Tong Univesity, 2008. |
| [14] | Li J F, Yan N Q, Qu Zan, et al. Catalytic Oxidation of Elemental Mercury over the Modified Catalyst Mn/α-Al2O3 at Lower Temperatures[J]. Environmental Science & Technology, 2010, 44 (1): 426-431. |
| [15] | 张安超, 郑雯雯, 向军, 等. 负载型MnOx/Al2O3催化剂低温下脱除烟气中单质汞特性[J]. 煤炭学报, 2013, 38 (S2): 471-477. |
| [16] | Dranga B A, Lazar L, Koeser H. Oxidation Catalysts for Elemental Mercury in Flue Gases-A Review [J]. Catalysts, 2012, 2 (1): 139-170. |
| [17] | Shen Z M, Ma J, Mei Z J, et al. Metal chlorides loaded on activated carbon to capture elemental mercury [J]. Journal of Environmental Sciences, 2010, 22 (11): 1814-1819. |
| [18] | Kong F H, Qiu J R, Liu H, et al. Catalytic oxidation of gas-phase elemental mercury by nano-Fe2O3 [J]. Journal of Environmental Sciences, 2011, 23 (4): 699-704. |
| [19] | Shan W P, Liu F D, He H, et al. An Environmentally-Benign CeO2-TiO2 Catalyst for the Selective Catalytic Reduction of NOx with NH3 in Simulated Diesel Exhaust[J]. Catalysis Today, 2012, 184 (1): 160-165. |
| [20] | Lee S M, Park K H, Hong S C. MnOx/CeO2-TiO2 Mixed Oxide Catalysts for the Selective Catalytic Reduction of NO with NH3 at Low Temperature[J]. Chemical Engineering Journal, 2012, 195-196 : 323-331. |
| [21] | Ji L, Sreekanth P M, Smirniotis P G, et al. Manganese Oxide/Titania Materials for Removal of NOx and Elemental Mercury from Flue Gas [J]. Energy & Fuels, 2008, 22 (4): 2299-2306. |
| [22] | Kijlstra W S, Biervliet M, Poels E K, et al. Deactivation by SO2 of MnOx/Al2O3 catalysts used for the selective catalytic reduction of NO with NH3 at low temperatures [J]. Applied Catalysis B: Environmental, 1998, 16 (4): 327-337. |
| [23] | Krishnakumar B, Niksa S. Predicting the impact of SO3 on mercury removal by carbon sorbents[J]. Proceedings of the Combustion Institute, 2011, 33 (2): 2779-2785. |
| [24] | Xu W Q, He H, Yu Y B. Deactivation of a Ce/TiO2 Catalyst by SO2 in the Selective Catalytic Reduction of NO by NH3 [J]. The Journal of Physical Chemistry C, 2009, 113 (11): 4426-4432. |
| [25] | Li Y, Murphy P D, Wu C Y, et al. Development of Silica/Vanadia/Titania Catalysts for Removal of Elemental Mercury from Coal-Combustion Flue Gas [J]. Environmental Science & Technology, 2008, 42 (14): 5304-5309. |
| [26] | Jin R B, Liu Y, Wu Z B, et al. Low-temperature selective catalytic reduction of NO with NH3 over Mn—Ce oxides supported on TiO2 and Al2O3: A comparative study [J]. Chemosphere, 2010, 78 (9): 1160-1166. |