 2015, Vol. 36
2015, Vol. 36



2. 西南大学计算机与信息科学学院, 重庆 400715;
3. 西南大学三峡库区生态环境教育部重点实验室, 重庆 400715
2. College of Computer and Information Science, Southwest University, Chongqing 400715, China;
3. Key Laboratory of Eco-environments in Three Gorges Reservoir Region, Ministry of Education, Southwest University, Chongqing 400715, China
沸石是一种被广泛应用于环境保护、 化工催化、 轻工、 农业等领域的矿物,特别是在净化空气[1,2]、 除臭抗菌[3]、 水污染处理[4,5]等环境污染治理中有着巨大的应用潜力. 沸石具有较高的比表面积和表面电荷密度,使其具有较强的离子吸附和交换性能. 大量研究发现[6, 7, 8, 9, 10],沸石对不同离子的吸附具有选择性,沸石的这种选择性吸附作用使之具有广阔的应用前景. 例如,人们根据天然斜发沸石对NH+4、 Cs+、 K+的较强选择性吸附,去除污水中的NH+4[7,8]、 放射性废液中的137Cs[9]以及从海水中提取K元素[10]等.
沸石对离子的选择性吸附是由沸石与不同离子间相互作用能的差异造成的,具体表现为离子吸附能力的差异. 经典理论认为高价离子的吸附能力强于低价离子,吸附能力服从Schulze-Hardy规则[11]. 但是,大量实验发现,沸石对同价离子的吸附同样存在差异,而这种差异称为离子特异性效应,又称Hofmeister效应. 近年来该效应日益引起物理学、 生物学领域专家的高度关注[12],并认为离子特异性效应蕴藏着重要的科学基础,并认为其重要性如同Mendel的豌豆杂交实验对于遗传学的重要性一样[13,14]. 同样,研究沸石的离子吸附和交换过程中的离子特异性对于更好地利用沸石具有重要意义.
离子特异性效应产生的主要原因仍然不够清楚[15],目前提出的主要原因可归纳为:表面络合作用[16]、 离子的体积效应[17]、 水合作用[18, 19, 20]、 诱导力[17]以及色散力[17, 21, 22]等. Ogawa等[23]研究发现,离子与沸石表面的色散力、 经典诱导力、 离子间相互作用以及共价作用都远远低于离子与沸石表面的静电作用,说明离子与沸石表面的相互作用以静电作用为主. 而且,这些理论也难以合理地解释离子交换平衡中的特异性效应[24],原因在于这些理论忽略了一个重要因素,就是水溶液条件下,带电颗粒表面存在着很强的电场,这一强电场对离子特异性效应具有重要影响. Liu等[25]发现,在蒙脱石表面发生的离子交换平衡中,离子在强电场中的极化差异是离子选择性交换的本质原因. 然而,沸石对不同离子吸附所表现出的特异性效应是否也是源于其表面电场的影响?电场影响离子极化的程度有多大?对于这些问题,人们目前并不清楚. 因此,本文以天然斜发沸石为实验材料,研究了不同组合的4种碱金属离子在沸石中交换平衡的特异性效应,以期为应用沸石通过离子交换吸附处理水体系中的目标离子提供理论支撑. 1 材料与方法 1.1 实验材料
本实验所采用的天然斜发沸石来自辽宁葫芦岛市康华科技有限公司. 用BET法测出其比表面积为32.677 m2 ·g-1. 实验试剂氯化钠、 氯化钾、 氯化锂和氯化铯均为分析纯,成都市科龙化工试剂厂生产. 1.2 实验方法 1.2.1 样品的前处理
Na/K饱和样的制备:分别称取100 g天然斜发沸石样品放入两个大锥形瓶中,加入0.2 mol ·L-1 的NaCl和KCl溶液1 000 mL,在25℃恒温下振荡24 h,离心,去除上清液,再次加入等量的0.2 mol ·L-1 的NaCl和KCl溶液1 000 mL,总共重复上述操作4次. 然后加入纯水,混合振荡24 h,离心,去上清液,重复处理2次. 最后将制好的钠饱和样和钾饱和样在70℃下烘干至恒重,过0.25 mm筛,混匀装袋备用. 1.2.2 离子交换平衡实验
设计了4组不同离子组合的交换平衡实验,有Li-Na、 Cs-Na、 Li-K和Cs-K交换,每个组合采用批处理实验.
分别称取约1 g K饱和样和Na饱和样于100 mL三角瓶中,向其中加入浓度梯度为0.001~0.1 mol ·L-1 的LiCl和CsCl溶液50 mL,密封,在恒温振荡器中298 K温度下平衡48 h. 然后离心,收集上清液,用火焰光度计测定溶液中的各离子浓度. 1.3 计算方法 1.3.1 离子交换吸附量的计算
交换性离子i的吸附量(cmol ·kg-1)计算公式如下:

被交换离子j吸附量(cmol ·kg-1)的计算公式如下:

测得了离子交换平衡时的离子浓度与平衡吸附量,则可根据下式计算离子的选择系数



选择系数能很好地反映离子的吸附能力的差异,方程(3)中,当Ki/j>1时,表示i离子的吸附能力强于j离子,当Ki/j<1时,表示i离子的吸附能力弱于j离子. 2 结果与讨论 2.1 碱金属离子在沸石表面交换平衡的离子特异性
在各组离子交换平衡实验结果中,以Li-Na、 Cs-Na、 Li-K和Cs-K离子交换达到平衡时的Li+和Cs+的吸附量与平衡时的交换性离子浓度做图,结果见图 1.
 | 图 1 交换性阳离子的吸附量与平衡浓度关系
Fig. 1 Relationship between the adsorption amount and the
equilibrium concentrations of the exchangable cations
|
从图 1可以看出,随着离子浓度的增加,交换离子的吸附量逐渐增加,但增加的趋势逐渐平缓,说明交换逐渐达到完全. Li+和Cs+交换Na+达到平衡时,天然斜发沸石对Li+、 Cs+的吸附量有显著差异. 例如当平衡浓度均为0.01 mol ·L-1 时,Li+和Cs+的平衡吸附量分别为23 cmol ·kg-1和112 cmol ·kg-1. 另外,Li/Na和Cs/Na交换体系中,Li+、 Cs+都存有一个最大吸附量,其中,Cs+的最大吸附量为180 cmol ·kg-1,是Li+最大吸附量(40 cmol ·kg-1)的4.5倍. 这表明天然斜发沸石的Na饱和样对Li+、 Cs+两种离子的选择顺序为Cs+>Li+. 在Li/K和Cs/K交换体系中,达到平衡时,两种阳离子的吸附量也具有显著差异. 同样以平衡浓度为0.01 mol ·L-1 来比较,Cs+的平衡吸附量为95 cmol ·kg-1,而Li+的平衡吸附量只有约15 cmol ·kg-1. 相同浓度的同种离子交换K+和Na+时,吸附量也是前者小于后者,说明K+在沸石表面的吸附能力强于Na+,导致离子交换平衡时,各离子的吸附量减小. K饱和样对Li+和Cs+离子的最大吸附量分别为35 cmol ·kg-1和110 cmol ·kg-1,Cs+的最大吸附量是Li+的3.14倍. 这表明天然斜发沸石的K饱和样对Li+、 Cs+的选择顺序为Cs+>Li+. 天然斜发沸石对这几种碱金属离子的吸附存在很强的选择性. 各体系中选择系数与离子强度的关系如图 2所示.
 | 图 2 选择系数与离子强度关系
Fig. 2 Relationship between the selectivity coefficient and the ion strength
|
Liu等[27]获得了经典的Poisson-Boltzmann方程在混合电解质体系下的解析解,结合Li等[28]对双电层平均浓度的定义,可得1 ∶1型电解质体系下表面电位与离子交换平衡的关系:

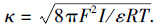 .
.由方程(6)、 (7)可得选择系数的理论表达式为:

由于x=1/κ处电位很小且φ1/κ→0,因此ZFφ0~ZFκ(φ0-φ1/κ)(1/κ)=ZFE(1/κ)= (C)可看成是离子在扩散层中的平均库仑能. 因此,上式可改写成:

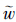 (C)为离子的平均库伦能. 从方程(9)可以看出离子的选择系数是由离子-表面库伦能决定的.
(C)为离子的平均库伦能. 从方程(9)可以看出离子的选择系数是由离子-表面库伦能决定的. 根据方程(8),可得同价离子之间的选择系数应该为1. 但是由图 2的实验结果可以看出,不同组合的离子交换平衡的选择系数都不是一个常数,而是随着离子强度的增加而减小的,其中KNa/Li>1、 KCs/Na>1、 KK/Li>1和KCs/K>1,结合4种交换体系的吸附量与平衡浓度的关系(图 1),可得天然斜发沸石对Li、 Na、 K、 Cs这4种碱金属离子的选择顺序为Cs+>K+>Na+>Li+.
上述4种碱金属离子在天然斜发沸石表面的选择性吸附顺序也就是离子特异性序列表明:离子与沸石表面的相互作用除了库仑能之外还存在着其他附加能量,尤其是在低电解质浓度下这种附加能更强. 接下来讨论这种附加能量是否来源于目前人们考虑的几种主要作用力,经过分析发现答案是否定的. 2.2 沸石离子交换中特异性效应的经典解释 2.2.1 表面络合作用
天然斜发沸石是一类含水的架状硅铝酸盐矿物,其表面电荷主要来源于同晶置换产生的永久负电荷[29],以及少量的表面羟基型功能基团[30]. 由于表面可变电荷数量较少,而且本实验是在较低离子强度下进行的,体系pH约为中性,故忽略氢离子对可变电荷的影响. 虽然沸石对重金属离子存在络合作用[16],但是碱金属离子的离子势较重金属离子小,发生表面水解而产生表面络合的可能性降低[31]. 碱金属离子与沸石结构中含有孤对电子的氧之间的共价作用远远低于静电能[23],说明共价作用也不是交换选择性的主要来源. 2.2.2 离子半径和水合作用
天然斜发沸石的有效孔径为0.4 nm左右[32],4种碱金属离子的直径均小于0.4 nm,说明离子半径不是影响其离子选择交换性的原因. Pauley[33]和Shainberg等[34]认为水化半径大的离子意味着离子的中心电荷距离黏土表面的距离越远,因此黏土-离子之间的静电力相互作用就比较弱,从而导致水化半径大的离子在表面的选择性就小[35,36]. 从表 1可以看出4种碱金属离子的水化半径大小为:Li+>Cs+>Na+>K+,当考虑水化半径时,其孔径对4种离子的筛分顺序应为:K+>Na+>Cs+>Li+. 水化半径大的离子,其水化能越高,离子就更倾向于留在溶液中,而不是被吸附[37]. 都与实验结果不符. 另一方面,近年来国内外很多研究[38, 39, 40, 41, 42]指出离子体积(包括水化体积)只在高浓度下才变得重要,而在稀溶液中离子的体积效应是可以忽略的. 因此,随着电解质浓度的降低,实验值与理论值之间的差异也应减小,与图 2的实验结果变化趋势相反. 甚至有人认为离子吸附在表面可能并不发生水化[17],因为在表面1.5 nm之内存在很强的水合斥力,水分子在表面被强烈极化[43]而变成非游离态. 说明天然斜发沸石对4种碱金属离子交换吸附中的强离子特异性效应并非来源于离子体积和水合作用.
| 表 1 Li+、 Na+、 K+和Cs+的离子半径、 水合半径、 水化能、 固有极化率和核外电子分布 Table 1 Ionic radius,hydrated radius,hydration free energy,static polarizability and extranuclear electron distribution of Li+,Na+,K+ and Cs+ |
如果离子交换平衡中强的离子特异性来源于色散力,方程(9)中指数项总能量除了库仑能还需加上色散能,即:

如果图 2中K与理论值1之间的差异来源于色散力,那么KC+D=K. 碱金属离子与表面的库仑能相等,即i(C)=j(C),所以方程(10)可以写成:

由于色散能不依赖于表面电位[45],可看成一个常数. 那么理论预测结果应为一个大于1的常数. 而且,研究发现色散力只在0.1 mol ·L-1 的高浓度下才起作用[46,47],也就是说随着电解质浓度的降低,选择系数的实验值越来越趋近于1. 显然这不能解释图 2中的实验结果,色散力仍然不是沸石中离子选择性的主要原因. 2.2.4 经典的离子诱导力
考虑诱导力之后的选择系数为:

i(I)表示离子i在扩散层中的诱导能.
离子i与j经典诱导能i(I)和j(I)的差可表示为:

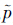 i(I)是离子i在扩散层中的平均偶极矩,
i(I)是离子i在扩散层中的平均偶极矩,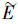 是双电层中的平均电场强度.
是双电层中的平均电场强度.

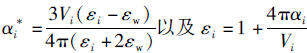 [44],αi*是离子在水溶液中的有效极化率,εi和εw是离子i与水分子的介电函数,ε0为真空中介电常数,Vi表示离子体积,αi离子固有极化率.
[44],αi*是离子在水溶液中的有效极化率,εi和εw是离子i与水分子的介电函数,ε0为真空中介电常数,Vi表示离子体积,αi离子固有极化率.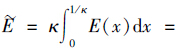
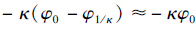 ,式中,1/κ为双电层厚度,φ0为表面电位,φ1/κ为1/κ处的电位. 将方程(13)、 (14)代入(12),可得经典诱导理论的选择系数:
,式中,1/κ为双电层厚度,φ0为表面电位,φ1/κ为1/κ处的电位. 将方程(13)、 (14)代入(12),可得经典诱导理论的选择系数:

考虑经典诱导能时,根据交换性离子的交换平衡结果,按下式计算表面电位:

从图 3可以看出,经典诱导能对各个体系中离子选择性吸附的贡献非常小. 考虑经典诱导能的选择系数仍然接近1,远远偏离实验值,理论预测与实验测定值的差异随离子组成不同而不同. 例如,在Li-Na和Li-K交换中,它们的差异随着表面电位的降低而增大,也就是说在高电位作用下,离子间选择性吸附差异更小; 在Cs-Na和Cs-K交换中的趋势正好相反. 上述结果表明经典诱导能大大低估了离子超极化率而不能正确描述离子交换平衡的界面行为. Ogawa等[23]研究发现沸石对碱金属离子和碱土金属离子的吸附能中,静电能是最主要的,极化作用,色散力,排斥力和共价作用等远远低于静电能而可忽略.
 | 图 3 K(C+I)与K的比较
Fig. 3 Comparison between K(C+I) and K
|
从前面的分析可以看出,表面络合作用,离子大小,水合作用、 色散力和经典诱导能均不能解释沸石离子交换平衡中的离子选择性. Liu等[24,25]认为吸附在双电层中的离子处于颗粒表面电荷产生的强电场中,其外层电子云的量子涨落必然受到强电场的诱导作用而发生极化,极化后的反离子本身也会屏蔽该处的电场,这种离子因量子涨落而产生的极化与表面电荷产生的强电场之间的相互作用简称为“电场-量子涨落”耦合作用. 本实验中天然斜发沸石在不同离子组合中的离子交换量也在35~180 cmol(-) ·kg-1之间(图 1),其表面电场强度范围为14~71 V ·nm-1. 这么强的电场说明在沸石表面同样存在这种“电场-量子涨落”耦合作用.
考虑离子-表面相互作用中“电场-量子涨落”耦合的极化能时,选择系数理论表达式为:

图 3的结果表明,经典的诱导理论强烈低估了离子的超极化率,因为它忽略了沸石表面大量电荷形成的强电场作用,故经典理论中的极化作用产生的偶极矩公式不适用于强电场中.
根据本课题组已有研究[25],离子与表面的静电能主要为库仑能与被电场放大了的极化能,它们与交换平衡时离子在本体溶液与扩散层中的分配的关系为:

根据以上方程组即可计算出各个体系中两种指示离子的偶极矩差以及体系表面电位的值.
从图 4可以看出,表面电位随着离子强度的增加而降低,这是因为高离子强度条件下,双电层被压缩,导致反离子屏蔽电荷能力增强,表面电位降低. 尽管在0.1 mol ·L-1 离子强度条件下,表面电位仍然较高,说明沸石表面电荷密度大.
 | 图 4 表面电位φ0与离子强度关系
Fig. 4 Relationship between the surface potential and the ion strength
|
另一方面,吸附在沸石表面的离子受到表面电场的强烈诱导,离子间偶极矩差与表面电位的关系如图 5所示.
 | 图 5 扩散层中离子偶极矩差与表面电位的关系
Fig. 5 Relationship between the ion dipole difference and the surface potential
|
从图 5可以看出,扩散层中偶极矩差随着表面电位的升高而增大,说明表面电场对离子外层电子云的结构具有重要影响,在研究离子与带电表面相互作用时,电场因素是不能被忽略的. 由于离子间外层电子排布的差异,在电场中的极化作用不同. 这种极化差异必然导致固/液界面上离子间交换吸附能力的差异. Cs+的外层电子云最为柔软,K+、 Na+次之,Li+最小,所以4种碱金属离子发生强极化作用的顺序为:Cs+>K+>Na+>Li+. 表面电位越高,电场越强,离子间极化差异就越大,导致偶极矩差越大. 所以,用“电场-量子涨落”耦合理论能很好地解释图 2中的实验现象. 在较低电位条件下,离子间偶极矩差减小至约等于0. 在其他黏土矿物如蒙脱石,伊利石表面不曾发现这种现象,原因在于沸石有许多的微孔,导致饱和样中吸附的被交换离子在低电位时存在一个很高的势垒而很难被交换下来,未被交换下来的离子被认为是不可交换的,所以偶极矩差异减弱,甚至出现反转. 这正好和图 1中的交换平衡结果相吻合,在不同的离子组合,不同浓度条件下,沸石的平衡吸附量存在着明显的特异性效应.
将方程(18)、 (19)代入(17),得到:

将图 4和图 5中的拟合曲线代入上式,即可获得考虑强电场作用后的选择系数与表面电位的经验预测关系曲线,结果如图 6所示. 吸附了离子的颗粒表面,其表面电位受本体溶液中离子强度的影响,高浓度下表面电位降低(图 4); 吸附在颗粒表面的离子,其极化作用受到表面电位的强烈影响,高表面电位下离子间偶极矩差越大(图 5); 选择系数随着表面电位的增加而增大,说明离子的交换吸附平衡的确是受到电场强度的驱动作用(图 6).
 | 图 6 选择系数与表面电位φ0的关系
Fig. 6 Relationship between the selectivity coefficient and the surface potential
|
从以上分析可以看出,离子交换吸附的特异性效应都可以归因于沸石表面电位的大小. 因此,在离子交换平衡中,凡是影响表面电位的因素均是离子存在选择性吸附的原因,本研究发现离子在表面电场中的极化能是根本原因. 同时,虽然离子体积效应不是引起表面选择性吸附的主要原因,但是由于沸石表面电荷密度高,其体积效应对离子在双电层中的分布也起着比较重要的作用[27],导致碱金属离子在沸石表面的交换平衡结果在高表面电位(绝对值大于0.2 V)下才表现出较大选择性差异,与蒙脱石上的交换平衡结果[25]存在一定差异.
值得一提的是,本研究选取的是碱金属离子在沸石表面的交换平衡,是因为碱金属离子结构较为简单,且同为一价离子,便于分析. 但是它对研究沸石处理水环境中重金属污染、 修复土壤以及氮磷流失等有着重要的指导意义,同时这也是进一步要研究的内容. 3 结论
(1) 天然斜发沸石表面的碱金属离子交换平衡存在着明显的离子特异性效应,其效应随着电解质浓度的降低而增大. 交换吸附选择性大小不能用经典的离子体积、 离子水化、 色散力、 诱导力以及表面络合等作用进行完整解释,而是来源于表面电场与离子量子涨落的耦合作用.
(2) 沸石表面的强电场对吸附离子的极化具有强烈的放大效应. 这种极化差异才是碱金属离子在沸石表面出现选择性吸附的根本动力.
(3) 通过分析发现,经典理论中的体积效应,色散效应在高电荷密度的沸石表面对“电场-量子涨落”耦合作用也具有较重要的影响,加上沸石的结构的复杂性,导致离子在沸石表面与蒙脱石、 伊利石表面交换平衡的差异.
| [1] | Aguado S, Polo A C, Bería M P, et al. Removal of pollutants from indoor air using zeolite membranes[J]. Journal of Membrane Science, 2004, 240 (1-2): 159-166. |
| [2] | Bhatia S, Abdullah A Z, Wong C T. Adsorption of butyl acetate in air over silver-loaded Y and ZSM-5 zeolites: Experimental and modelling studies[J]. Journal of Hazardous Materials, 2009, 163 (1): 73-81. |
| [3] | Ziolek M, Sobczak I, Dudzik A, et al. Application of modified zeolites and mesoporous materials for deodorization[J]. Studies in Surface Science and Catalysis, 2008, 174 (Part A): 555-560. |
| [4] | 李德生, 黄晓东, 王占生. 生物沸石反应器在微污染水源水处理中的应用[J]. 环境科学, 2000, 21 (5): 71-73. |
| [5] | 柳萍, 王建龙. 天然沸石在水污染控制中的应用[J]. 离子交换与吸附, 1996, 12 (4): 378-382. |
| [6] | 张铨昌, 杨华蕊, 韩成. 天然沸石离子交换性能及其应用[M]. 北京: 科学出版社, 1986. 1-78. |
| [7] | 张曦, 吴为中, 温东辉, 等. 生物沸石床污水脱氮效果及机理[J]. 环境科学, 2003, 24 (5): 75-80. |
| [8] | 赵统刚, 吴德意, 陈建刚, 等. 粉煤灰合成沸石同步脱氨除磷特性的研究[J]. 环境科学, 2006, 27 (4): 696-700. |
| [9] | Abusafa A, Yücel H. Removal of 137Cs from aqueous solutions using different cationic forms of a natural zeolite: clinoptilolite[J]. Separation and Purification Technology, 2002, 28 (2): 103-116. |
| [10] | Ames L L. Some zeolite equilibria with alkaline earth metal cations[J]. The American Mineralogist, 1964, 49 (3): 35-39. |
| [11] | Hiemenz P C, Rajagopalan R. Principles of colloid and surface chemistry[M]. New York: Marcel Dekker, 1977. 588-592. |
| [12] | López-León T, Santander-Ortega M J, Ortega-Vinuesa J L, et al. Hofmeister effects in colloidal systems: influence of the surface nature[J]. The Journal of Physical Chemistry C, 2008, 112 (41): 16060-16069. |
| [13] | Lo Nostro P, Ninham B W. Hofmeister phenomena: an update on ion specificity in biology[J]. Chemical Reviews, 2012, 112 (4): 2286-2322. |
| [14] | Kunz W, Lo Nostro P, Ninham B W. The present state of affairs with Hofmeister effects[J]. Current Opinion in Colloid & Interface Science, 2004, 9 (1-2): 1-18. |
| [15] | Hashemi M, Chasteen T G. Hofmeister effect challenge[J]. Analytical and Bioanalytical Chemistry, 2011, 400 (3): 643-644. |
| [16] | Zong P F, Wang H, Pan H, et al. Application of NKF-6 zeolite for the removal of U(VI) from aqueous solution[J]. Journal of Radioanalytical and Nuclear Chemistry, 2013, 295 (3): 1969-1979. |
| [17] | Parsons D F, Boström M, Nostro P L, et al. Hofmeister effects: interplay of hydration, nonelectrostatic potentials, and ion size[J]. Physical Chemistry Chemical Physics, 2011, 13 (27): 12352-12367. |
| [18] | Tielrooij K J, Garcia-Araez N, Bonn M, et al. Cooperativity in ion hydration[J]. Science, 2010, 328 (5981): 1006-1009. |
| [19] | Nucci N V, Vanderkooi J M. Effects of salts of the hofmeister series on the hydrogen bond network of water[J]. Journal of Molecular Liquids, 2008, 143 (2-3): 160-170. |
| [20] | Peula-García J M, Ortega-Vinuesa J L, Bastos-González D. Inversion of Hofmeister series by changing the surface of colloidal particles from hydrophobic to hydrophilic[J]. The Journal of Physical Chemistry C, 2010, 114 (25): 11133-11139. |
| [21] | Zhang Y J, Cremer P S. Chemistry of Hofmeister anions and osmolytes[J]. Annual Review of Physical Chemistry, 2010, 61: 63-83. |
| [22] | Ninham B W, Duignan T T, Parsons D F. Approaches to hydration, old and new: Insights through Hofmeister effects[J]. Current Opinion in Colloid & Interface Science, 2011, 16 (6): 612-617. |
| [23] | Ogawa K, Nitta M, Aomura K. A theoretical study of the site selectivity of the zeolite cation. 1. Site selectivities of alkali and alkaline earth metal cations in zeolite A [J]. The Journal of Chemical Physics, 1978, 82 (14): 1655-1660. |
| [24] | Liu X M, Li H, Li R, et al. Strong non-classical induction forces in ion-surface interactions: General origin of Hofmeister effects[J]. Scientific Reports, 2014, 4: 5047. |
| [25] | Liu X M, Li H, Du W, et al. Hofmeister effects on cation exchange equilibrium: quantification of ion exchange selectivity[J]. The Journal of Physical Chemistry C, 2013, 117 (12): 6245-6251. |
| [26] | Davies C W. Ion association[M]. London: Butterworths, 1962. 37-53. |
| [27] | Liu X M, Li H, Li R, et al. Generalized poisson—boltzmann equation taking into account ionic interaction and steric effects[J]. Communications in Theoretical Physics, 2012, 58 (3): 437-440. |
| [28] | Li H, Wu L S. On the relationship between thermal diffusion and molecular interaction energy in binary mixtures[J]. The Journal of Physical Chemistry B, 2004, 108 (36): 13821-13826. |
| [29] | Ćurkovi DćL, Cerjan-Stefanovi DćŠ, Filipan T. Metal ion exchange by natural and modified zeolites[J]. Water Research, 1997, 31 (6): 1379-1382. |
| [30] | 吴大清, 刁桂仪, 彭金莲, 等. 矿物界面作用与环境工程材料[J]. 矿物岩石地球化学通报, 1998, 17 (4): 217-223. |
| [31] | 武玫玲. 土壤矿质胶体的可变电荷表面对重金属离子的专性吸附[J]. 土壤通报, 1985, 16 (2): 89-94. |
| [32] | Rajic N, Stojakovic D, Daneu N, et al. The formation of oxide nanoparticles on the surface of natural clinoptilolite[J]. Journal of Physics and Chemistry of Solids, 2011, 72 (6): 800-803. |
| [33] | Pauley J L. Prediction of cation-exchange equilibria[J]. Journal of the American Chemical Society, 1954, 76 (5): 1422-1425. |
| [34] | Shainberg I, Kemper W D. Hydration status of adsorbed cations[J]. Soil Science Society of America Journal, 1966, 30 (6): 707-713. |
| [35] | Driesner T, Seward T M, Tironi I G. Molecular dynamics simulation study of ionic hydration and ion association in dilute and 1 molal aqueous sodium chloride solutions from ambient to supercritical conditions[J]. Geochimica et Cosmochimica Acta, 1998, 62 (18): 3095-3107. |
| [36] | Feng H J, Zhou J, Lu X H, et al. Communication: Molecular dynamics simulations of the interfacial structure of alkali metal fluoride solutions[J]. The Journal of Chemical Physics, 2010, 133 (6): 061103. |
| [37] | Tansel B. Significance of thermodynamic and physical characteristics on permeation of ions during membrane separation: Hydrated radius, hydration free energy and viscous effects[J]. Separation and Purification Technology, 2012, 86: 119-126. |
| [38] | Borukhov I, Andelman D, Orland H. Steric effects in electrolytes: A modified Poisson-Boltzmann equation[J]. Physical Review Letters, 1997, 79 (3): 435. |
| [39] | Laird D A, Shang C. Relationship between cation exchange selectivity and crystalline swelling in expanding 2: 1 phyllosilicates[J]. Clays and Clay Minerals, 1997, 45 (5): 681-689. |
| [40] | Liu X M, Li H, Li R, et al. A new model for cation exchange equilibrium considering the electrostatic field of charged particles[J]. Journal of Soils and Sediments, 2012, 12 (7): 1019-1029. |
| [41] | Lou P, Lee J Y. Ionic size effect on the double layer properties: a modified poisson-boltzmann theory[J]. Bulletin of the Korean Chemical Society, 2010, 31 (9): 2553-2556. |
| [42] | Teppen B J, Miller D M. Hydration energy determines isovalent cation exchange selectivity by clay minerals[J]. Soil Science Society of America Journal, 2006, 70 (1): 31-40. |
| [43] | Pashley R M, Israelachvili J N. DLVO and hydration forces between mica surfaces in Mg2+, Ca2+, Sr2+, and Ba2+ chloride solutions[J]. Journal of Colloid and Interface Science, 1984, 97 (2): 446-455. |
| [44] | Parsons D F, Ninham B W. Importance of accurate dynamic polarizabilities for the ionic dispersion interactions of alkali halides[J]. Langmuir, 2010, 26 (3): 1816-1823. |
| [45] | Boström M, Williams D R M, Ninham B W. Specific ion effects: why DLVO theory fails for biology and colloid systems[J]. Physical Review Letters, 2001, 87 (16): 168103. |
| [46] | Johnson S B, Scales P J, Healy T W. The binding of monovalent electrolyte ions on α-alumina. I. Electroacoustic studies at high electrolyte concentrations[J]. Langmuir, 1999, 15 (8): 2836-2843. |
| [47] | Kosmulski M. Confirmation of the differentiating effect of small cations in the shift of the isoelectric point of oxides at high ionic strengths[J]. Langmuir, 2002, 18 (3): 785-787. |