 2014, Vol. 35
2014, Vol. 35



2. 中国科学院烟台海岸带研究所, 烟台 264003
2. Yantai Institute of Coastal Zone Research, Chinese Academy of Sciences, Yantai 264003, China
第二次世界大战战后至今,在全球各地均发现大量化学武器埋藏点,其中部分化学武器经长期腐蚀,其内部有毒有害物质泄漏到自然环境中,二苯砷酸是其中较为常见的砷污染物[1,2]. 它是二苯氯砷或二苯氰砷的水解或氧化产物,后两者常被用于制造致喷剂和致呕剂[3]. 经研究发现,DPAA能长期稳定地存在于自然环境中. DPAA同土壤颗粒的结合能力较弱,具有较强的向下迁移能力,使得泄漏地区土壤与地下水环境的砷污染现象严重,严重威胁当地居民的身体健康[4, 5]. 因此,亟需开发出能快速有效处理该污染物的修复技术.
目前对于处理DPAA污染的方法研究较少,主要包括生物修复、 客土置换和化学催化降解[2]. 日本科学家曾筛选出能直接降解DPAA的微生物[6],其中Nakamiya等[7]发现的菌株对DPAA的降解率可达到90%以上,但需要指出,实验中DPAA的初始浓度仅为5 mg ·L-1,远低于实际污染水平. 迄今未发现能降解较高浓度DPAA的微生物. Guan等[8]发现通过调控土壤环境中的硫还原性,能加强土著微生物对DPAA的转化,但所需时间较长. Nakajima等[9]也曾利用双氧水催化降解DPAA,但反应条件需高温,也制约了该方法的实际应用. 纳米二氧化钛光催化剂凭其强大的催化能力和便利的反应条件已被广泛应用于降解有机污染物,并取得良好效果[10]. 利用二氧化钛处理砷污染也是目前的研究重点,前人研究表明二氧化钛能有效催化毒性较高的亚砷酸离子向砷酸离子转化[11],对于甲基砷、 二甲基砷和苯砷酸等有机砷也有较强的降解效果[12, 13, 14],但尚无关于利用二氧化钛催化降解DPAA的报道.
本实验室前期研究发现,添加二氧化钛的土壤除能提高对DPAA的固定能力外,经紫外灯照射,能快速降解DPAA,且最终产物为砷酸离子. 为了进一步了解二氧化钛对DPAA的降解过程及其影响因素,本实验研究了二氧化钛对DPAA的降解动力学,主要考察DPAA初始浓度和吸附过程对该反应动力学的影响. 除进一步研究盐离子强度和酸碱度等环境条件变化对催化降解DPAA反应速率的影响外,还研究不同活性氧基团在催化反应过程中的作用. 通过对催化反应过程的充分了解,以期为后续的实际应用打下理论基础.
1 材料与方法 1.1 供试材料
二苯砷酸购自Wako Co.,纯度为97%. 催化剂二氧化钛为Degussa P25,由80%锐钛矿型与20% 金红石型的二氧化钛组成. 超氧化物歧化酶(SOD)活性约为3 500 unit ·mg-1,相对分子质量约为32 500,购自Fisher Scientific Inc. 乙腈为色谱纯,其余试剂均为分析纯. 1.2 吸附实验
DPAA在二氧化钛表面的吸附实验采用批平衡法. DPAA浓度包括5、 10、 15、 20、 25和30 mg ·L-1. 背景溶液为1 mmol ·L-1 NaNO3,二氧化钛浓度为 0.1 g ·L-1. 经超声处理30 min后,磁力搅拌1 h已达到吸附平衡. DPAA在氧化钛表面的吸附量Qads按下式计算:
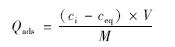
式中,ci为DPAA的初始浓度,ceq为达到吸附平衡时上清液中DPAA的浓度. V为溶液体积 (mL),M为吸附剂质量 (g).
吸附等温线用Langmuir单表面方程拟合,该方程表示如下:
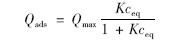
上式经变换后得:
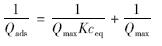
对 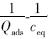 做图,可计算出最大吸附量Qmax和吸附平衡常数K.
1.3 光催化实验
做图,可计算出最大吸附量Qmax和吸附平衡常数K.
1.3 光催化实验
采用Photochem-3型光化学反应仪(北京纽比特公司)进行光催化降解实验,光源为500 W高压汞灯. 反应液置于Pyrex玻璃试管中,反应液中心处平均光辐射强度(200~500 nm)为42 mW ·cm-2. 若无其他说明,反应溶液中DPAA的浓度为20 mg ·L-1,二氧化钛浓度为0.1 g ·L-1,离子强度为1 mmol ·L-1 NaNO3,pH值为4.5.
在光照前,所有反应溶液均在超声仪中超声30 min,以便分散溶液中的二氧化钛,随后在遮光条件下磁力搅拌1 h,使氧化钛对DPAA的吸附达到平衡. 光解实验中,同时设置暗对照,在相同条件下考察DPAA的光降解情况.
在光催化反应前及反应中,向反应溶液中通入氮气或氧气,以研究溶解氧对二氧化钛光催化降解二苯砷酸的影响. 向反应溶液中添加NaHCO3(200 mg ·L-1 HCO-3)、 NaN3(200 mg ·L-1 N-3)、 SOD(4 000 unit ·mL-1)及EDTA ·2Na(500 mg ·L-1)以研究不同活性氧基团在光催化降解二苯砷酸反应中的作用.
分别在0、 5、 10、 30、 60和90 min中取样,每次取1 mL,经0.22 μm滤膜过滤,待测DPAA浓度. 以上每组实验均重复3次. 1.4 样品分析
使用HPLC(岛津)检测DPAA浓度. 检测条件为: SPD-m20A检测器,岛津VP-ODS C18色谱柱(4.6 mm× 150 mm,5 μm),柱温30℃; 流动相为19 ∶81的乙腈与0.02 mol ·L-1 KH2PO4溶液(磷酸调节pH=2.7),进样量为10 μL,流速为1 mL ·min-1,检测波长为220 nm. DPAA的保留时间为10.72 min.
2 结果与分析 2.1 暗反应中二氧化钛P25对二苯砷酸的吸附特征
二氧化钛光致催化反应主要发生于催化剂表面,因此其对反应物的吸附特征会直接影响后续的催化降解反应[15]. 试验考察了P25对二苯砷酸的吸附特征. 吸附等温线采用Langmuir单表面方程拟合,结果如图 1所示.
 | 误差线为一倍标准偏差,下同 图 1 DPAA在二氧化钛表面的吸附等温线 Fig. 1 Langmuir isotherm adsorption of DPAA onto TiO2 photocatalyst |
由图 1可见,该方程能很好地拟合二苯砷酸在二氧化钛表面的吸附等温线,拟合系数R2为0.985. 其中,平衡常数K为0.10 L ·mg-1,最大吸附量Qmax约为(5.0±0.2)×103 mg ·kg-1. 前人研究结果发现甲基砷、 二甲基砷和苯砷酸在P25表面的最大吸附量分别为1.2×104、 5.1×103和2.5×104 mg ·kg-1 [12, 13]. 本实验中,DPAA与DMA在催化剂表面Qmax相同,而PAA与MMA等单一取代基有机砷的Qmax则要远高于相对应的双取代基有机砷. 这可能是由有机砷的分子结构不同所致. 分子结构较大的苯基取代砷会产生较大的空间位阻,降低该类有机砷在吸附剂表面的吸附能力; 而双取代基则导致砷化合物结构上羟基数目的下降,减少了该类有机砷可供吸附剂表面吸附的活性基团. 二者均阻碍了吸附反应:吸附剂—OH+HO—As—R 吸附剂—O—As—R+H2O [16].
此外,根据Langmuir单表面方程的假设前提以及P25二氧化钛表面的羟基密度(4.8 ·OH nm2)[17],理论上可以推算该纳米颗粒对DPAA的最大吸附量约为1.0×105 mg ·kg-1,而实验值仅为理论值的5%,Zheng等[12]的研究发现PAA在P25催化剂表面的实际吸附量也仅有理论值的30%. 除了有机砷分子结构产生空间位阻效应外,也可能是由于二氧化钛在水溶液中会很快发生团聚现象,其比表面积降低,导致部分羟基结合位点被屏蔽,从而使得有机砷吸附量下降. 2.2 二氧化钛光催化降解二苯砷酸的非均相反应动力学
二氧化钛光催化反应为非均相反应过程,其初始阶段反应动力学多符合一级反应动力学[18]. 其中反应动力学常数k的大小可以表征反应速率的快慢.
图 2为不同浓度二氧化钛光催化降解DPAA的反应动力学,反应动力学常数k分别为k0.1 g ·L-1=1.47×10-2 min-1、 k1.0 g ·L-1=1.91×10-2 min-1和k2.0 g ·L-1=5.94×10-2 min-1. 表明反应速率随着二氧化钛使用浓度的增加而加快. 此时,二氧化钛对DPAA的吸附量也随着P25浓度的增加而提高,据此推测吸附过程会影响催化降解反应.
 | 图 2 不同浓度二氧化钛对DPAA降解的反应动力学
Fig. 2 Effects of different dosages of TiO2
on the photo-catalytical degradation kinetics
|
Langmuir-Hinshelwood反应动力学模型将反应过程分为催化反应与吸附两个过程,可进一步表征催化剂表面及表面吸附特性对二氧化钛光催化反应的影响[19]. 该方程如下所示:
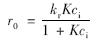
式中,r0为反应刚开始时反应速率,kr为反应速率常数,K为吸附平衡常数,ci为DPAA初始浓度.
将上式变换可得:
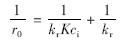
对 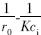 做图,可计算求出kr和吸附平衡常数K.
做图,可计算求出kr和吸附平衡常数K.
利用该反应模型笔者研究了不同浓度DPAA对二氧化钛催化降解的影响. 结果如图 3所示.
 | 图 3 纳米二氧化钛催化降解二苯砷酸的Langmuir-Hinshelwood动力学方程
Fig. 3 Langmuir-Hinshelwood plot of TiO2 photocatalytic degradation of DPAA
|
根据图 3求出L-H反应动力学模型各参数并列于表 1. 与其他有机砷相比,DPAA的反应速率最快,且苯基取代砷的反应速率均显著高于甲基取代砷. 这可能是由于两类有机砷化合物在催化反应过程中的反应机制不同所致.
| 表 1 有机砷在吸附等温线及Langmuir-Hinshelwood反应动力学模型中的主要参数 Table 1 Main parameters of organic arsenics in adsorption isotherm and Langmuir-Hinshelwood model |
二氧化钛光催化反应中主要涉及羟基自由基对有机物的氧化[20]. 前人研究发现,苯基取代的有机砷与羟基自由基的反应包括苯环的羟基化[12],而甲基取代的有机砷与羟基自由基的反应则涉及甲基上的脱氢反应[14]. 前者的反应速率[~1010 mol ·(L ·s)-1]要远高于后者[~108 mol ·(L ·s)-1][18]. 由此可以推测, DPAA分子结构上具有两个苯环,与羟基自由基的结合位点要多于PAA,从而导致其反应速率远高于其他有机砷.
此外,苯基取代砷在两个模型中的平衡常数基 本相同,而甲基取代砷的吸附平衡常数在两个模型中却极不相同. 有研究将后者归因于光辐射过程中氧化钛表面结合位点的变化; 反应产物对吸附位点的竞争结合以及小分子砷化合物在吸附剂中的双层吸附特征[21, 22]. 由此推断,在光催化反应前后,DPAA在二氧化钛表面的吸附过程受外界因素影响较小,均适用于Langmuir吸附等温线. 进一步推断,当DPAA浓度较高时,其催化反应的主要产物无机砷酸离子可以竞争结合氧化钛表面的吸附位点,进而影响DPAA在氧化钛表面的吸附及降解反应速率. 2.3 离子强度及pH值对二氧化钛光催化降解二苯砷酸速率的影响
二氧化钛光催化反应包括吸附与光反应两个过程,因此先研究不同离子强度及酸碱条件下,二氧化钛对DPAA吸附量的变化. 结果如图 4所示. 从中可见,二氧化钛对DPAA的吸附量受离子强度变化的影响较小,离子强度最小时,吸附量略高; 当pH值从3变为4.5时,吸附量小幅上升,当pH>4.5时,吸附量随pH升高而降低.
 | 图 4 pH值及离子强度对氧化钛吸附DPAA的影响
Fig. 4 Effects of different pH value and ionic strengths on the adsorption of DPAA by TiO2
|
上述现象可能是由于DPAA专性吸附于吸附剂表面所致[5]. 离子强度不影响专性吸附,因此实验中DPAA的吸附量受离子强度的影响较小. 离子强度较高时,导致催化剂颗粒发生快速团聚,表面羟基数目下降,导致对DPAA的吸附量降低. DPAA的pKa值为5.2[23],随着溶液pH值上升,水溶液中的DPAA由中性分子转变为阴离子. 同时二氧化钛的等电点(PZC)为6.8,酸性条件下,二氧化钛表面的正电荷随pH减小而增加; 碱性条件下,二氧化钛表面负电荷随pH升高而增加. 当pH值约为5.2时,DPAA与二氧化钛表面带异种电荷,存在静电吸附,导致吸附量增高; 当pH值下降时,DPAA为中性分子,与二氧化钛表面不存在静电引力; 而在碱性条件下,DPAA则与二氧化钛表面存在静电斥力,二者均导致吸附量下降.
离子强度及pH变化对二氧化钛光催化降解DPAA速率的影响如图 5所示. 从中可见,当离子强度最小时,光催化反应速率最大; 当pH值从3变为4.5时,反应速率变快,当pH>4.5时,反应速率则随pH升高而下降. 离子强度及pH值对光催化反应速率影响的趋势与二者对二氧化钛吸附DPAA的影响趋势相一致,可以推测催化剂表面对DPAA的吸附量会影响光反应速率的快慢. 此外,在强酸或强碱环境中,二氧化钛在水溶液中的稳定性会增加[24],且由于氧化钛表面正或负电荷的增加有利于光生电子或空穴向催化剂表面转移,会增强催化效率[25]. 但本实验中在强酸或强碱环境中,光反应速率反而较低,甚至当pH=11时,基本无催化反应发生,这也表明二氧化钛光催化降解DPAA的反应主要受催化剂表面吸附DPAA的影响.
 | 图 5 不同离子强度与pH值下的二氧化钛催化降解DPAA的反应动力学
Fig. 5 Time profiles of DPAA degradation in the TiO2 suspension at various ionic strength and pH value
|
二氧化钛光催化过程中可能发生的反应方程如下所示[19]:
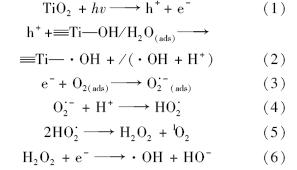
这一系列反应中涉及到溶液中的溶解氧. 因此,笔者研究了溶解氧对二氧化钛催化降解DPAA反应速率的影响,结果如图 6所示. 从中可见,增加溶解氧的浓度能提高催化降解速率,而降低溶解氧的浓度则导致反应速率下降. 这可能是由于光生电子e-被溶解氧捕获,降低了电子/空穴对的复合,提高反应效率. 其次,溶解氧浓度的增加也可能会提高反应体系中各活性氧基团的生成量,如羟自由基和单线态氧等[26, 27].
 | 图 6 溶解氧对二氧化钛降解DPAA反应动力学的影响
Fig. 6 Effect of dissolved oxygen on the photo-catalytical kinetics of DPAA
|
由上述反应方程式(1)~(7)可知,该反应可能涉及羟自由基( ·OH)、 超氧阴离子(O ·-2)和单线态氧(1 O2)等. 通过研究各种活性氧基团在二氧化钛光催化降解DPAA的反应中所起作用,可进一步推测该反应机制. 为此,利用活性氧基团猝灭剂,研究各猝灭剂对DPAA降解速率快慢的影响,进而判断各活性氧基团在反应过程中所做贡献. 各猝灭剂对活性氧基团的猝灭反应列于表 2.
| 表 2 活性氧基团猝灭剂反应 Table 2 Quenching Reactions of Scavengers |
各淬灭剂对DPAA降解速率的影响如图 7所示,从中可见,EDTA对催化反应影响最大,反应速率由对照组的9.1×10-3 min-1降为5.0×10-4 min-1,反应基本停滞; 其次分别为NaHCO3和NaN3,表观反应速率分别下降至kNaHCO3=3.4×10-3 min-1和kNaN3=6.1×10-3 min-1; 而SOD则对反应速率无影响.
 | 图 7 不同活性氧基团猝灭剂对降解反应动力学的影响
Fig. 7 Time profiles of PA degradation in the TiO2 suspension with various scavengers
|
由反应方程(2)可知,氧化钛表面存在带电空穴是生成 ·OH的前提条件,而EDTA作为h+的猝灭剂,导致 ·OH无法生成,这表明 ·OH可能是氧化钛催化降解DPAA的主要活性氧基团. NaHCO3和NaN3与 ·OH反应可减少体系中 ·OH,降低反应速率. 尽管 NaN3还可同时与1 O2反应,如若1 O2参与催化降解DPAA,则NaN3对该反应的抑制程度应高于仅同 ·OH相反应的NaHCO3. 但实验结果显示NaN3对降解DPAA的抑制程度却小于NaHCO3,这表明,催化降解DPAA的过程中1 O2所起作用要小于 ·OH. 由于SOD对降解反应基本无影响,可以推测O ·-2 在该反应中贡献也较小.
综上所述,二氧化钛光催化降解DPAA的反应过程可由图 8表示.
 | 图 8 纳米二氧化钛催化降解二苯砷酸的反应途径
Fig. 8 Proposed photo-degradation pathway of DPAA by nano-TiO2
|
二氧化钛在紫外线激发下,其表面形成电子/空穴对,经一系列催化反应形成羟基自由基,并随后氧化已吸附在氧化钛表面的DPAA,并将其降解为无机砷酸根离子.
3 结论
(1)本研究发现二氧化钛光催化剂能快速、 有效地降解DPAA,其反应过程可分为吸附与光反应两个阶段,且DPAA在催化剂表面吸附量的大小会影响光反应速率. 整个光催化反应过程可以用L-H动力学方程很好地拟合. 其中在光照前后,DPAA在P25二氧化钛表面的吸附过程均符合Langmuir单表面吸附等温线.
(2)各种环境因素如盐离子强度和酸碱度的变化均会影响降解速率. 其中离子强度通过改变纳米颗粒粒径大小,降低其稳定性,继而引起纳米颗粒量子尺寸效应的改变,最终导致反应速率下降. 而酸碱度则通过纳米颗粒对DPAA吸附量的改变导致反应速率随吸附量的提高而加快.
(3)二氧化钛光催化降解DPAA的反应过程中羟基自由基贡献最大. 由于二氧化钛光催化技术已经被广泛应用于废水处理中,这表明利用二氧化钛光降解环境中的DPAA有较强的应用前景,但仍需进一步研究各种影响因素,如自然界中广泛存在的HCO-3或可溶性有机质(DOM)等羟自由基猝灭剂对该方法应用的影响.
| [1] | Hempel M, Daus B, Vogt C, et al. Natural attenuation potential of phenylarsenicals in anoxic groundwaters [J]. Environmental Science and Technology, 2009, 43 (18): 6989-6995. |
| [2] | Daus B, Hempel M, Wennrich R, et al. Concentrations and speciation of arsenic in groundwater polluted by warfare agents [J]. Environmental Pollution, 2010, 158 (11): 3439-3444. |
| [3] | Leermakers M, Baeyens W, De Gieter M, et al. Toxic arsenic compounds in environmental samples: Speciation and validation [J]. Tractrends in Analytical Chemistry, 2006, 25 (1): 1-10. |
| [4] | Maejima Y, Murano H, Iwafune T, et al. Adsorption and mobility of aromatic arsenicals in Japanese agricultural soils [J]. Soil Science and Plant Nutrition, 2011, 57 (3): 429-435. |
| [5] | Wang A A, Li S X, Teng Y, et al. Adsorption and desorption characteristics of diphenylarsenicals in two contrasting soils [J]. Journal of Environmental Sciences, 2013, 25 (6): 1172-1179. |
| [6] | Harada N, Takagi K, Baba K, et al. Biodegradation of diphenylarsinic acid to arsenic acid by novel soil bacteria isolated from contaminated soil [J]. Biodegradation, 2010, 21 (3): 491-499. |
| [7] | Nakamiya K, Nakayama T, Ito H, et al. Degradation of arylarsenic compounds by microorganisms [J]. FEMS Microbiology Letters, 2007, 274 (2): 184-188. |
| [8] | Guan L, Hisatomi S, Fujii K, et al. Enhanced transformation of diphenylarsinic acid in soil under sulfate-reducing conditions [J]. Journal of Hazardous Materials, 2012, 241-242: 355-362. |
| [9] | Nakajima T, Kawabata T, Kawabata H, et al. Degradation of phenylarsonic acid and its derivatives into arsenate by hydrothermal treatment and photocatalytic reaction [J]. Applied Organometallic Chemistry, 2005, 19 (2): 254-259. |
| [10] | Chong M N, Jin B, Chow C W K, et al. Recent developments in photocatalytic water treatment technology: A review [J]. Water Research, 2010, 44 (10): 2997-3027. |
| [11] | Guan X H, Du J S, Meng X G, et al. Corrigendum to "Application of titanium dioxide in arsenic removal from water: A review" [J. Hazard. Mater. 215-216 (2012) 1-16]J]. Journal of Hazardous Materials, 2012, 221-222: 303. |
| [12] | Zheng S, Cai Y, OShea K E. TiO2 photocatalytic degradation of phenylarsonic acid [J]. Journal of Photochemistry and Photobiology A-Chemistry, 2010, 210 (1): 61-68. |
| [13] | Xu T L, Cai Y, OShea K E. Adsorption and photocatalyzed oxidation of methylated arsenic species in TiO2 suspensions [J]. Environmental Science and Technology, 2007, 41 (15): 5471-5477. |
| [14] | Xu Z H, Jing C Y, Li F S, et al. Mechanisms of photocatalytical degradation of monomethylarsonic and dimethylarsinic acids using nanocrystalline titanium dioxide [J]. Environmental Science and Technology, 2008, 42 (7): 2349-2354. |
| [15] | Fujishima A, Rao T N, Tryk D A. TiO2 photocatalysts and diamond electrodes [J]. Electrochimica Acta, 2000, 45 (28): 4683-4690. |
| [16] | Jing C Y, Meng X G, Liu S Q, et al. Surface complexation of organic arsenic on nanocrystalline titanium oxide [J]. Journal of Colloid and Interface Science, 2005, 290 (1): 14-21. |
| [17] | Mueller R, Kammler H K, Wegner K, et al. OH surface density of SiO2 and TiO2 by thermogravimetric analysis [J]. Langmuir, 2003, 19 (1): 160-165. |
| [18] | Xu T, Kamat P V, Joshi S, et al. Hydroxyl radical mediated degradation of phenylarsonic acid [J]. Journal of Physical Chemistry A, 2007, 111 (32): 7819-7824. |
| [19] | 邓南圣, 吴峰. 环境光化学[M]. 北京: 化学工业出版社, 2003. 363-373. |
| [20] | 韩世同, 习海玲, 史瑞雪, 等. 半导体光催化研究进展与展望 [J]. 化学物理学报, 2003, 16 (5): 339-349. |
| [21] | Xu T L, Kamat P V, OShea K E. Mechanistic evaluation of arsenite oxidation in TiO2 assisted photocatalysis [J]. Journal of Physical Chemistry A, 2005, 109 (40): 9070-9075. |
| [22] | Xu Y M, Langford C H. Variation of Langmuir adsorption constant determined for TiO2-photocatalyzed degradation of acetophenone under different light intensity [J]. Journal of Photochemistry and Photobiology A-Chemistry, 2000, 133 (1-2): 67-71. |
| [23] | Nakamiya K, Shibata Y, Ito H, et al. Synthesis of phenyl arsenic analytical standards related to contaminated well water in Kamisu, Ibaraki, Japan [J]. Applied Organometallic Chemistry, 2005, 19 (2): 282-286. |
| [24] | French R A, Jacobson A R, Kim B, et al. Influence of ionic strength, pH, and cation valence on aggregation kinetics of titanium dioxide nanoparticles [J]. Environmental Science and Technology, 2009, 43 (5): 1354-1359. |
| [25] | Wang P H, Yap P S, Lim T T. C-N-S tridoped TiO2 for photocatalytic degradation of tetracycline under visible-light irradiation [J]. Applied Catalysis A-General, 2011, 399 (1-2): 252-261. |
| [26] | Liang H C, Li X Z, Yang Y H, et al. Effects of dissolved oxygen, pH, and anions on the 2, 3-dichlorophenol degradation by photocatalytic reaction with anodic TiO2 nanotube films [J]. Chemosphere, 2008, 73 (5): 805-812. |
| [27] | Pelaez M, de la Cruz A A, OShea K, et al. Effects of water parameters on the degradation of microcystin-LR under visible light-activated TiO2 photocatalyst [J]. Abstracts of Papers of the American Chemical Society, 2011, 45 (12): 3787-3796. |
| [28] | Beltran F J, Garcia-Araya J F, Alvarez P M, et al. Aqueous degradation of atrazine and some of its main by-products with ozone hydrogen peroxide [J]. Journal of Chemical Technology and Biotechnology, 1998, 71 (4): 345-355. |
| [29] | Land E J, Prutz W A. Reaction of azide radicals with amino-acids and proteins [J]. International Journal of Radiation Biology, 1979, 36 (1): 75-83. |
| [30] | Minero C, Mariella G, Maurino V, et al. Photocatalytic transformation of organic compounds in the presence of inorganic ions. 2. Competitive reactions of phenol and alcohols an a titanium dioxide-fluoride system [J]. Langmuir, 2000, 16 (23): 8964-8972. |
| [31] | Klug D, Fridovic I, Rabani J. A direct demonstration of the catalytic action of superoxide dismutase through use of pulse radiolysis [J]. Journal of Biological Chemistry, 1972, 247 (15): 4839-4842. |