 2014, Vol. 35
2014, Vol. 35



随着我国经济的发展和城市化进程的加快,“退二进三”旧城改造及“退城进园”异地搬迁政策的实施,越来越多的地下水污染场地在土地再开发利用过程中暴露出来.由于当前我国污染地下水修复技术极为缺乏,因此,寻求廉价、 高效和绿色的快速修复技术已成为我国环境修复领域的研究热点和难点[1,2,3].
在各种地下水污染修复和治理技术中,原位化学氧化技术(in situ chemical oxidation,ISCO)是最为活跃的一个研究方向.根据2010年度美国EPA超级基金报告,采用ISCO修复土壤和地下水的案例呈明显上升趋势[4].其中,Fenton高级氧化技术具有原料易得、 反应条件温和、 反应快速等特点,已广泛应用于国内外废水处理以及有机污染土壤和地下水的原位修复中[5].然而,在对Fenton技术研究和应用过程中发现,Fenton仅在酸性条件下处理效果较好,中性或碱性条件易析出沉淀,限制了其推广应用; 其次,要使反应进行必须保证适量浓度Fe2+,过量的Fe2+会被自由基氧化,增加水中高价铁离子浓度,降低了处理效率,并消耗大量亚铁离子和H2 O2,导致处理费用增加.
综上,Fenton体系中Fe2+活化H2 O2的瓶颈为:过量Fe2+会快速地清除羟基自由基( ·OH),同时本身转化为Fe3+,即限速步骤是Fe3+返回Fe2+的反应.由于该反应速率常数较低,使反应过程铁离子循环受阻,体系铁离子将以Fe3+形式积累,最终导致有机污染物降解不完全.因此,保持溶液中适量的Fe2+对于产生 ·OH和提高目标污染物的氧化效率是极其重要的.
基于此,采用非均相的含铁催化剂(零价铁、 纳米零价铁、 铁氧化物等)替代Fe2+分解H2 O2从而引发Fenton反应,已逐渐引起了国内外研究人员的兴趣.与其它催化剂相比,nZVI具有比表面积大、 反应活性高的特点.近年来,随着nZVI售价降低,以nZVI催化H2 O2已成为污染场地地下水修复的新热点.此方法将nZVI还原和Fenton高级氧化技术有机结合起来,一方面克服了单独nZVI还原仅发生硝基还原或还原脱氯反应、 过程本身无矿化作用的缺点[6],另一方面也避免了某些污染物单独氧化难以进行、 易生成多种有毒中间产物的弊端[7, 8],使得污染物先还原后再深度脱毒和矿化过程得以快速实现.
目前,已有一些研究陆续开展了这方面的工作.伊朗Alizadeh等[9]尝试nZVI-Fenton修复BTEX污染地下水,发现pH 为3.0、 BTEX、 nZVI和H2 O2浓度分别为4、 9和150 mg ·L-1时,99%的BTEX可在15 min内降解; 同组的Shirazi等[10]也发现nZVI-Fenton工艺降解地下水中卡马西平效果良好,5 mg ·L-1的卡马西平可在5 min内快速分解.韩国Moon等[11]采用nZVI-Fenton耦合工艺可快速对0.3 mmol ·L-1橙色2号染料进行脱色和矿化,1 h内脱色率和矿化度分别达到95%和53%; 采用0.3348 g ·L-1的nZVI先还原后氧化的新型工艺处理浓度约100 mg ·L-1酸性黑24染料废水,仅需10 min即达完全脱色,1.5 h内TOC去除率高达93.9%[12].印度Babuponnusami等[13]通过研究光电助nZVI-H2 O2工艺降解苯酚,发现在pH 6.2、 nZVI 0.5 g ·L-1、 H2 O2 0.5 g ·L-1、 电流密度12 mA ·cm-2时,0.5 h内污染物达到完全去除,电解产生的H2 O2可为连续产生 ·OH创造条件.国内王晴晴等[14]也发现以自制纳米铁为催化剂可有效催化分解H2 O2去除水中2,4,6-三氯酚,优化实验条件下超过95%的三氯酚(浓度1.10 mmol ·L-1)在20 min内被降解完毕.Xu等[15]利用纳米铁催化分解H2 O2去除水中4-氯-3-甲基苯酚,初始条件为pH 6.1、 0.5 g nZVI和3.0 mmol ·L-1 H2 O2时,0.7 mmol ·L-1的污染物于15 min内分解完毕,1 h内TOC去除率高达63%.还有研究发现添加多金属氧酸盐和多金属钨酸盐能够促进系统内铁离子循环和液相H2 O2的生成,可进一步提高nZVI-H2 O2系统氧化能力[16].
可见,将nZVI还原转化和Fenton工艺进行耦合,有助于强化污染物的快速降解和矿化.中国城市化进程中对土地的迫切需求往往要求修复快速化,这为该类技术的应用提供了良好前景[17].而且,同西方发达国家相比,我国采用Fenton试剂降解有机污染物的实践起步较晚,且主要应用于化工、 印染、 医药和农药等行业排放的废水处理,在难降解有机污染土壤和地下水修复方面则鲜有报道[5].
地下水中4-氯硝基苯是一种稳定的高毒性污染物,目前对其修复研究极少.本研究通过建立nZVI催化H2 O2耦合系统,分析其修复某污染场地4-氯硝基苯污染地下水的性能和降解机制,探讨修复技术的可行性和有效性,以期为该污染场地地下水的快速修复提供科学依据和关键技术. 1 材料与方法 1.1 实验材料
本研究所用的4-氯硝基苯污染地下水取自某化工厂搬迁遗留场地,取样深度为-0.5 m,表观无色、 无味、 透明,pH 3.0,其各敏感组分平均含量如下:铁0.065 mg ·L-1、 锰0.468 mg ·L-1、 镁73.4 mg ·L-1、 钙63.9 mg ·L-1、 铜0.009 mg ·L-1、 铝0.11 mg ·L-1,有机污染物以4-氯硝基苯(4-ClNB)为主,平均浓度为60 mg ·L-1. 1.2 nZVI制备方法
nZVI颗粒通过“液相还原法”制得[18],具体方法为:将150 mL 0.2 mol ·L-1的FeSO4溶液倒入500 mL的三口平底烧瓶内,预先以200 mL ·min-1流量通入高纯氮气15 min,用滴液漏斗以20 mL ·min-1的速度缓慢加入150 mL 0.6 mol ·L-1的硼氢化钠溶液,同时持续通入高纯氮气,并保持机械搅拌.待硼氢化钠溶液滴加完成后,静置陈化30 min.反应完成后氮气保护下用真空泵抽滤,无水乙醇冲洗,真空干燥、 保存,备用. 1.3 实验方法
移取450 mL 4-氯硝基苯污染地下水转入500 mL的平底烧瓶内,加盖密封置于磁力搅拌器上.为模拟地下水厌氧环境,实验前污染地下水预先微曝氮气10 min,使其溶解氧低于0.5 mg ·L-1.投加既定质量的nZVI,开启磁力搅拌器,转速400 r ·min-1,然后通过加入定量的H2 O2触发反应,开始计时.每隔一定时间用注射器采样.在室温下取样时,加入1滴自由基清除剂叔丁醇到样品中,停止反应.样品通过0.45 μm滤膜后进行测试分析.
在进行反应中间产物捕捉时,为较为全面地捕获中间产物,将反应过程分割为两个阶段,即nZVI还原过程和后续Fenton反应过程,并将时间适当延长,减缓反应速率.具体而言:先投加既定质量的nZVI,反应一段时间,取样完毕后再投加一定浓度的H2 O2,再进行取样,分别进行前处理后进行GC/LC-MS和离子色谱(IC)分析. 1.4 分析方法
目标有机污染物采用HPLC测定:地下水样经Sigmal-13型离心机10000 r ·min-1离心10 min,取0.6 mL上清液与0.9 mL甲醇充分混合,经0.22 μm有机滤膜过滤后进行分析.检测仪器为:Waters1525高效液相色谱仪(Milford MA,美国)、717自动进样器、 2487双波长紫外检测器.分析条件为:ZORBAX Eclipse XDB-C18分离柱(4.6 mm×150 mm,5 μm),流动相为甲醇/0.02% H3PO4 6/4,流速1.0mL ·min-1,进样体积10 μL,柱温30℃. 4-氯苯胺检测波长为230 nm,其余有机污染物检测波长为254 nm.
将实验制备的nZVI利用超声波分散,制成电镜试样,通过Philip XL-30-ESEM扫描电子显微镜(SEM)在15 kV下扫描; 粒径采用Omnisorp100CX型物理吸附仪(Coulter,美国)测定.地下水中金属含量采用X系列微波消解电感耦合等离子体质谱法(ICP-MS,Thermo Elemental,美国)进行分析.阴离子和小分子有机酸采用美国Dionex-1000型离子色谱(IC)测定,AG11-HC色谱柱(4.0 mm×250 mm),电导检测器,KOH淋洗液(流速1.0 mL ·min-1,30mmol ·L-1),进样体积25 μL.
中间产物采用GC-MS(Agilent 6890N/5975B,美国)和LC-MS(Agilent1100,美国)测定,100mL样品采用甲基叔丁基醚萃取,回收有机相加适量无水硫酸钠脱水后浓缩至体积1 mL后进行分析.GC-MS为EI源,轰击电压70 eV,HP-5MS柱(30 m×0.25 mm i.d.×0.25 μm),不分流,载气为高纯氦,流速1.0 mL ·min-1,初温50℃(2 min),10℃ ·min-1升至150℃(1 min),之后20℃ ·min-1升至270℃(5 min),进样口温度250℃,接口温度280℃.LC-MS在Agilent 1100 LC/MSD SL上进行; 离子源为ESI,正/负离子扫描,扫描范围50~600 nm,碎片电压100 V; 干燥气流速12 L ·min-1,雾化器压力为50 PSI,干燥温度350℃,毛细管电压为4000 V. 2 结果与分析 2.1 nZVI颗粒的表征
nZVI颗粒的扫描电镜如图 1所示.从中可以看出,nZVI大部分成球形或椭球形,粒径经测定分布在10~50nm,同时发现颗粒有不同程度的黏连,这是由于磁性nZVI受地磁力、 粒子间的静磁力及表面张力共同作用发生了团聚.此外,新制备的nZVI暴露在空气中会打火自燃,表明其具有较高的活性. nZVI X射线衍射图如图 2所示.通过与JCPDF标准数据卡片的数据对照,图 2中所示的峰(44.85°)为零价纳米铁的衍射峰,表明材料中所含的铁为单质铁,即为零价铁的特征峰[19, 20].
 | 图 1 nZVI的扫描电镜谱图 Fig.1 SEM photograph of NZVI |
 | 图2 NZVI X射线衍射 Fig.2 XRD pattern of nZVI |
在30℃、 pH 3.0、 nZVI投加浓度448 mg ·L-1条件下,考察了H2 O2浓度(0~19.6 mmol ·L-1)对污染地下水中4-ClNB的降解影响,结果如图 3所示.
 | 图3 H2 O2浓度对污染地下水中4-ClNB去除的影响 Fig.3 Influence of H2 O2 concentration on the degradation of 4-ClNB in contaminated groundwater |
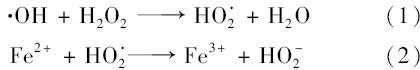
初始H2 O2投加量4.90 mmol ·L-1、 污染地下水初始pH值3.0、 温度30℃条件下,不同nZVI投加量对地下水中4-ClNB去除率的影响如图4所示. 从中可知,当nZVI投加量分别为268.8 mg ·L-1和358.4 mg ·L-1时,在30 min时,地下水中4-ClNB的降解率可达100%; 当nZVI投加量分别为89.6 mg ·L-1和179.2 mg ·L-1时,在35 min后去除率亦可达100%.总的来看,随着nZVI投加量的增加(0~358.4 mg ·L-1),4-ClNB降解的拟一级反应速率常数由0.011 min-1增至0.1778 min-1,nZVI投加量与4-ClNB的拟一级反应速率常数之间存在良好的线性关系(如图 5),相关系数R2>0.92.这可能是因为,当nZVI投加量增大时,其吸附性能增强,反应过程主要为先吸附再降解,4-ClNB吸附到nZVI表面的反应及非反应位点,反应速率较快.当nZVI投加量相对减少时,其吸附性能降低,由此可见4-ClNB浓度的降低主要是由降解作用引起的.
 | 图4 nZVI投加量对污染地下水中4-ClNB去除的影响 Fig.4 Influence of nZVI addition on the degradation of 4-ClNB in contaminated groundwater |
 | 图5nZVI投加量与地下水中4-ClNB拟一级反应降解速率常数的相关性 Fig.5 Correlation between nZVI addition and pseudo-first order reaction rate constant of 4-ClNB in contaminated groundwater |
图6为不同初始pH值下nZVI催化H2 O2体系对地下水中4-ClNB的去除情况.初始pH为2.0、 3.0时分别在12 min和20 min内4-ClNB去除率达到100%,初始pH分别为4.0、 5.0和7.0时30 min内其去除率分别下降为约50%、30%和30%左右,而初始pH为11.0时不发生反应,30min内4-ClNB基本无去除.由此可以看出,酸性条件有利于体系反应活性的提高,地下水中4-ClNB的去除率随着地下水初始pH的增加逐渐降低.其原因可能是因为在酸性条件下, ·OH是占优势的自由基.在碱性条件下, ·OH会发生如式(3)的反应,由此生成的 ·O2H氧化性远不如 ·OH高[22],反应速率减慢.初始pH值会影响地下水中Fe2+离子的存在形式,随着地下水pH值的升高,Fe2+离子的存在形式发生反应式(4)中的变化,从而影响 ·OH的生成速率和产生量,使得催化剂量减少或失去活性.此外,pH值较高时会抑制反应式(5)的反应,使生成的 ·OH数量减少.较高的pH值还会使H2 O2发生无效分解,分解为O2和H2 O,从而影响H2 O2的利用率,使4-ClNB的降解率降低.当地下水pH值由2.0逐渐升高至11.0时,nZVI还原反应生成的铁离子会转化为氢氧化亚铁沉淀,其附着在nZVI表面,阻碍电子的转移,并抑制反应的进行,从而导致4-ClNB的降解率大幅下降[23].综上,4-ClNB在酸性条件下较大pH范围内都能得到有效降解转化.
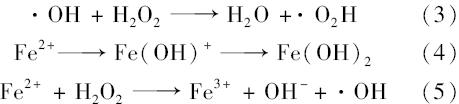
 | 图6 地下水初始pH值对4-ClNB去除的影响 Fig.6 Influence of initial pH on the degradation of 4-ClNB in contaminated groundwater |
应用化学技术进行地下水污染治理和修复时,地下水中一些无机阴离子常会对反应产生影响.图7分别比较了投加1 mmol ·L-1的Cl-、 NO-3和SO2-4离子对nZVI-Fenton耦合工艺降解4-ClNB行为的影响.从中可知,反应体系投加一价的Cl-和NO-3离子,30 min 内4-ClNB的降解几乎无影响; 而投加二价的SO2-4离子会使4-ClNB的降解率下降10%左右.这与Shirazi等[10]的研究结论一致.推测可能的原因是:SO2-4离子倾向吸附于nZVI表面,占据了nZVI部分表面活性位点,降低了 ·OH生成数量进而导致了4-ClNB降解率的下降; 而一价Cl-和NO-3离子相对于二价SO2-4离子水溶性更佳,吸附于nZVI表面的机率更低,因而一价Cl-和NO-3离子对体系4-ClNB降解率的影响较二价的SO2-4离子小.
 | 图7 投加阴离子对4-ClNB去除的影响 Fig.7 Influence of anion addition on the degradation of 4-ClNB in contaminated groundwater |
为了进一步阐明nZVI催化H2 O2强化转化降解地下水中4-ClNB的途径,分别应用GC/MS、 LC/MS和IC技术分析了反应过程的主要中间产物.4-ClNB转化产物GC/MS分析在tR分别为2.390、 7.897、 10.32和18.27 min检测到产物峰,应用NIST05质谱数据库进行比对,发现其与对苯醌、 4-氯亚硝基苯、 4-氯苯胺和4-氯偶氮苯匹配率分别达91%、 94%、 95%和95%.应用LC/MS分析时分别在tR为2.235、 3.305、 11.112和12.58 min检测到质荷比(m/z)144.1、 128.0、 108.1和267.0的负离子峰,初步判定其可能为对氯羟基苯胺(m=144)、 对氯苯胺(m=128)、 对苯醌(m=108)和对氯氧化偶氮苯(m=267),具体见表1. 从中可知,nZVI催化H2 O2复合体系形成的中间产物既包括还原产物(对氯亚硝基苯、 对氯羟基苯胺、 对氯氧化偶氮苯、 对氯偶氮苯、 对氯苯胺),也包括上述产物的深度氧化产物,如对苯醌、 小分子羧酸(乙酸、 甲酸、 草酸)和氯离子等.
| 表1 nZVI催化H2 O2降解污染地下水4-ClNB过程中检测到主要产物 Table 1 Detected intermediates during 4-ClNB transformation by the nZVI catalytic hydrogen peroxide process |
在已有报道ZVI还原转化硝基芳烃过程中,苯环上硝基经一系列加氢取代依次转化为亚硝基、羟胺基和氨基,同时其中少量中间产物可发生偶氮反应生成偶氮苯类物质后再通过加氢作用转化为芳香胺[24, 25].生成的芳香胺产物通过Fenton氧化可进一步降解或矿化.据此,结合实验过程检测到的中间产物,提出nZVI催化H2 O2体系转化降解4-ClNB的建议性途径,具体如图8所示.4-ClNB在nZVI作用下还原为4-氯亚硝基苯、 4-氯羟基苯胺,进而还原为4-氯苯胺;此外,生成的过渡态产物4-氯亚硝基苯和4-氯羟基苯胺也可以发生反应生成4-氯氧化偶氮苯,其在产物4-氯苯胺参与下、 脱水生成4-氯叠氮苯,4-氯叠氮苯加氢、 均裂,最终分解为4-氯苯胺; 中间产物4-氯苯胺在 ·OH攻击下生成4-氯苯亚胺自由基、 经脱氯、 脱氨生成对苯醌,然后氧化开环、 进一步分解为小分子酸(甲酸、 乙酸、 草酸等)、 二氧化碳和水等.
 | 图8 nZVI催化H2 O2转化降解4-ClNB可能的反应途径 Fig.8 Proposed pathway of 4-ClNB degradation by the nZVI catalytic H2 O2 process |
(1)单独nZVI和过氧化氢均不能有效修复污染地下水中4-ClNB,而采用nZVI催化H2 O2可以快速降解地下水中4-ClNB.
(2)nZVI、 H2 O2投加量和污染地下水初始pH值对4-ClNB的降解均有较大影响.4-ClNB的降解符合拟一级反应动力学,其反应速率常数随nZVI投加量(0~358.4 mg ·L-1)的增加呈线性增大,随H2 O2投加量的增加先增大后减小,H2 O2最佳投加量为4.90 mmol ·L-1; 地下水初始pH值为酸性时,4-ClNB可得到有效降解.
(3)在初始pH值为3.0、 H2 O2浓度为4.90 mmol ·L-1、 nZVI投加量268.8 mg ·L-1、 温度30℃时,nZVI催化H2 O2工艺可在30 min内完全转化降解污染地下水中的4-ClNB.
(4)地下水中的Cl-、 NO-3在30 min内对nZVI-Fenton耦合工艺降解4-ClNB行为基本无影响,而SO2-4离子会使4-ClNB的降解率下降10%左右.因此,一价Cl-和NO-3离子对体系4-ClNB降解率的影响较二价的SO2-4离子小.
(5) GC/MS、 HPLC/MS和IC分析表明,nZVI催化H2 O2工艺降解4-ClNB的中间产物有4-氯亚硝基苯、 4-氯羟基苯胺、 4-氯氧化偶氮苯、 4-氯偶氮苯、 4-氯苯胺、 对苯醌、 甲酸、 乙酸、 草酸和氯离子等.
| [1] | Lapworth D J, Baeran N, Stuart M E, et al. Emerging organic contaminants in groundwater: a review of sources, fate and occurrence[J]. Environmental Pollution, 2012, 163 (2): 287-303. |
| [2] | 陈慧敏, 仵彦卿. 地下水污染修复技术的研究进展[J]. 净水技术, 2010, 29 (6): 5-8. |
| [3] | 赵勇胜. 地下水污染场地污染的控制与修复[J]. 吉林大学学报(地球科学版), 2007, 37 (2): 303-310. |
| [4] | USEPA 2010. Superfund Remedy Report, Thirteenth Edition (EPA-542-R-10-004). EPA. Office of Solid Waste and Emergency Response (OSWER). September, 2010. SRR 13th edition is available electronically at www. Clu-in.org/asr. |
| [5] | 崔英杰, 杨世迎, 王萍, 等. Fenton原位化学氧化法修复有机污染土壤和地下水研究[J]. 化学进展, 2008, 20 (7/8): 1196-1201. |
| [6] | Mantha R, Taylor K E, Biswas N, et al. A continuous system for Fe0 reduction of nitrobenzene in synthetic wastewater[J]. Environmental Science & Technology, 2001, 35 (15): 3231-3236. |
| [7] | Li Y P, Cao H B, Liu C M, et al. Electrochemical reduction of nitrobenzene at carbon nanotube electrode[J]. Journal of Hazardous Materials, 2007, 148 (1/2): 158-163. |
| [8] | Nishino S F, Spain J C. Degradation of nitrobenzene by a Pseudomonas pseudoalcaligenes[J]. Applied Environmental Microbiology, 1993, 59 (8): 2520-2525. |
| [9] | Alizadeh F M, Torabian A, Bidhendi G R N, et al. Fenton and photo-Fenton oxidation of petroleum aromatic hydrocarbons using nanoscale zero-valent iron[J]. Journal of Environmental Engineering-ASCE, 2013, 139 (7): 966-974. |
| [10] | Shirazi E, Torabian A, Bidhendi G N. Carbamazepine removal from groundwater: effectiveness of the TiO2/UV, nanoparticulate zero-valent iron, and Fenton (NZVI/H2O2) processes[J]. CLEAN-Soil, Air, Water, 2013, 41 (11): 1062-1072. |
| [11] | Moon B H, Park Y B, Park K H. Fenton oxidation of orange Ⅱ by pre-reduction using nanoscale zero-valent iron[J]. Desalination, 2011, 268 (1-3): 249-252. |
| [12] | Shu H Y, Chang M C, Chang C C. Integration of nanosized zero-valent iron particles addition with UV/H2O2 process for purification of azo dye Acid Black 24 solution[J]. Journal of Hazardous Materials, 2009, 167 (1-3): 1178-1184. |
| [13] | Babuponnusami A, Muthukumar K. Removal of phenol by heterogeneous photo electro Fenton-like process using nano-zero valent iron[J]. Separation and Purification Technology, 2012, 98 (1): 130-135. |
| [14] | 王晴晴, 武俐, 邰超, 等. 纳米铁催化过氧化氢降解2,4,6-三氯酚的研究[J]. 河南理工大学学报(自然科学版), 2011, 30 (2): 244-248. |
| [15] | Xu L, Wang J. A heterogeneous Fenton-like system with nanoparticulate zero-valent iron for removal of 4-chloro-3-methyl phenol[J]. Journal of Hazardous Materials, 2011, 186 (1): 256-264. |
| [16] | Mylon S E, Sun Q, Waite T D. Process optimization in use of zero valent iron nanoparticles for oxidative transformations[J]. Chemosphere, 2010, 81 (1): 127-131. |
| [17] | 谢剑, 李发生. 中国污染场地的修复与再开发的现状分析. 可持续发展-东亚及太平洋地区研究报告[R], 世界银行, 2010. |
| [18] | Wang C B, Zhang W X. Synthesizing nanoscale iron particles for rapid and complete dechlorination of TCE and PCBs[J]. Environmental Science & Technology, 1997, 31 (7): 2154-2156. |
| [19] | Shi L N, Zhang X, Lin Y M, et al. Synthesis, characterization and kinetics of bentonite supported nZVI for the removal of Cr(Ⅵ) from aqueous solution[J]. Chemical Engineering Journal, 2011, 171 (2): 612-617. |
| [20] | Zhang X, Lin S, Chen Z L, et al. Kaolinite-supported nanoscale zero-valent iron for removal of Pb2+ from aqueous solution: Reactivity, characterization and mechanism [J]. Water Research, 2011, 45 (11): 3481-3488. |
| [21] | 黄进. 天然水中典型阴离子对芬顿反应的影响[J]. 化学工程师, 2004, 108 (9): 3-6. |
| [22] | Rathi A, Rajor H K, Sharma R K. Photodegradation of direct yellow-12 using UV/H2O2/Fe2+ [J]. Journal of Hazardous Materials, 2003, 102 (2-3): 231-241. |
| [23] | 胡六江, 李益民. 有机膨润土负载纳米铁去除废水中硝基苯[J]. 环境科学学报, 2008, 28 (6): 1107-1112. |
| [24] | Agrawal A, Tratnyek P G. Reduction of nitro aromatic compounds by zero-valent iron metal[J]. Environmental Science & Technology, 1995, 30 (1): 153-160. |
| [25] | Klausen J, Ranke J, Schwarzenbach R P. Influence of solution comparsion and column aging on the reduction of nitroaromatic compounds by zero-valent iron[J]. Chemosphere, 2001, 44 (4): 511-517. |