 2014, Vol.35
2014, Vol.35 



臭氧是城市大气中的重要污染物,其主要由大气中挥发性有机物(VOC)与氮氧化物(NOx, x为1或2)在光照条件下反应产生. 在众多VOC中,芳香烃是城市大气中浓度较高的主要物种. 大气中芳香烃的排放占全球人为排放非甲烷烃的25%~30%[1],其主要来自汽车尾气排放和燃料溶剂的挥发损失[2, 3, 4]. 在机动车较多的城市,芳香烃所占据的百分比往往较高,如在北京和天津,芳香烃浓度在所有非甲烷烃中最高,分别达到58.2%和49.9%[5],在乡村地区,芳香烃仅为总VOC含量的约1.7%[6]. 大气中含量最高的芳香烃主要为苯、 甲苯、 二甲苯、 三甲苯、 乙苯和苯乙烯等,其含量占总芳香烃的60%~75%[7, 8,10]. 比较文献报道的各城市大气中芳香烃浓度,发现苯和乙苯浓度一般处于较高水平,表 1给出了近几年文献中报道的我国几个大城市及其它国家一些城市大气中的苯和乙苯的浓度.
| 表 1 各城市大气环境中苯和乙苯的浓度 /μg ·m-3Table 1 Concentrations of benzene and ethylbenzene in urban atmospheric environment/μg ·m-3 |
大部分芳香烃具有很高的O3生成活性. 有研究认为在英国,芳香烃对局地O3的贡献达30%[13],在北欧地区达40%[14]. 在北京和天津,芳香烃对O3生成潜势的贡献,可以分别占到总臭氧生成潜势的75%和73%[5]. 为了评价不同VOC的光化学反应活性,一般借助VOC与OH自由基的反应速率,以及VOC在光化学反应过程中对OH自由基的贡献和消耗. 前者称为VOC的动力学反应活性,后者称为VOC的机制反应活性. 常见的3种表征VOC光化学反应活性的方法有:①等效丙烯浓度法,该法是以某一VOC物种与OH自由基反应的速率与丙烯与OH自由基反应的速率常数之比求得等效丙烯浓度以评估该物种的光化反应能力. ②最大增量反应活性法(maximum incremental reactivity, MIR),Carter[15, 16]提出增量反应活性法(incremental reactivity,IR),该法是根据有NOx存在的光照体系中加入一定量VOC物种后所产生的6 h时微量O3浓度变化,来衡量单位VOC生成臭氧的能力. ③光化O3产生潜势(photochemical ozone creation potential,POCP),由Derwent等[13]发展,POCP值代表某碳氢化合物相对于乙烯而言,每单位排放量所产生的O3能力. 用POCP模拟时发现苯环类会有较高的POCP值[17]. 目前仅有Kelly等[18]通过设计烟雾箱实验,直接测量了苯醛、 苯、 甲苯以及对二甲苯的8 h IR值,发现结果与Carter[15, 16]的MIR值比较一致. 而对于乙苯的光氧化过程还缺少相应烟雾箱实验数据的支持.
现有MIR值均由SAPRC(Statewide Air Pollution Research Center)机制[19]模拟得到,由于其为归纳机制,忽略了一些中间反应过程,因而由该机制得到的可能与其它类型机制得到的臭氧增量反应活性存在差异,特别是已有的反应机制中,还缺少对乙苯光氧化过程的烟雾箱实验数据验证. MCM(master chemical mechanism)为特定机制,由英国Leeds大学发展[20,21],其主要优点是给出了每个物种的详细降解过程,有助于人们更加深入地了解大气化学过程的内在机制,因而用其模拟VOC臭氧生成活性可能更加合理. 目前还没有基于MCM得到的MIR值. 另外一些环境因子对O3生成潜势影响的研究还较少,如MIR值是在低湿条件下获得的,真实大气中高湿度对MIR结果有何影响仍需要研究.
本研究使用自制的烟雾箱,分析了苯和乙苯光氧化生成臭氧的过程,并比较了MCM机制对苯和乙苯光氧化实验的模拟情况,最后在烟雾箱实验的基础上通过MCM计算了以上两个物种的臭氧生成活性.
苯(北京化学试剂公司,纯度为99.5%),乙苯(北京国药化学试剂公司,纯度为98.5%),NO2(北京海普气体有限公司, 101×10-6,稀释气为高纯N2),实验背景空气采用合成空气(由80%高纯N2与20%高纯O2组成,湿度低于5%).
实验中先将一定量的NO2标准气引入反应器,然后用合成空气将气化后的芳香烃引入反应器. 为使反应物在反应器中得到充分混合,先将所有反应物静止1 h,再测量各反应物的初始浓度,然后打开模拟光源和风扇并开始计时,以后每隔1 h测量O3和NOx(NO, NO2)浓度. 芳香烃采用长光程FTIR分析,测量前0.5 h用高纯N2对仪器内部进行吹扫,以减少H2 O和CO2的影响. NOx (NO,NO2) 采用Model 42C 氮氧化物分析仪分析,O3采用Model 49C 臭氧分析仪分析,CO采用Model 48C一氧化碳分析仪分析.
表 2为苯和乙苯-NOx光氧化实验的初始条件,实验1~9为苯-NOx光氧化体系,实验10~27为乙苯-NOx光氧化体系,其中实验1和实验2,以及实验10与实验11用于验证烟雾箱的可重复性.
| 表 2 苯和乙苯-NOx光氧化体系的反应初始条件 Table 2 Initial experimental conditions for benzene-NOx and ethylbenzene-NOx irradiations |
烟雾箱实验结果的可重复性是衡量VOC-NOx光氧化实验质量的重要参照. 为了表征烟雾箱的可重复性,分别做了苯和乙苯光氧化体系的两组重复实验. 这里以乙苯-NOx光照体系为例(实验10与实验11),两组反应物NO、 NO2和乙苯的初始浓度几乎相等,温度和湿度也非常接近,其中反应过程中温度分别为301 K和300 K,湿度均小于5%,因此由反应初始条件引起的偏差可以忽略. 实验10与实验11中臭氧、 NO和NO2时间廓线的结果比较见图 1. 发现在6 h的反应过程中,两组实验中的臭氧以及NOx的时间变化曲线几乎完全重合,其中整个反应过程中臭氧的最大偏差为4%,NO相差小于1×10-9,由于反应后期NO浓度接近仪器检测线,此时算NO的偏差已经没有意义,NO2的最大相差仅3×10-9,偏差为18%. 苯-NOx光氧化实验结果也类似,其中8 h实验中,臭氧值最大仅相差8.6%. 由以上比较可以看出:本研究使用的烟雾箱体系在苯-NOx和乙苯-NOx光氧化实验过程具有很好的重复性.
 | 图 1 乙苯-NOx光氧化重复性实验
Fig.1 Reproducibility experiments of ethylbenzene-NOx irradiations
|
 | 图 2 不同湿度时MCM机制对苯光氧化的模拟 Fig.2 MCM simulations of benzene-NOx irradiations under different RH conditions |
 | 图 3 不同湿度时MCM机制对乙苯光氧化的模拟
Fig.3 MCM simulations of ethylbenzene-NOx irradiations under different RH conditions
|
其它不同C6H6/NOx比率时,MCM对苯光氧化的模拟结果与实验结果的偏差与实验4和实验9情况类似,即干燥时MCM模拟的臭氧峰值浓度比实验值偏高约20%,高湿时MCM模拟的臭氧峰值比实验值偏高约40%. 受检测条件及实验时间等因素制约,烟雾箱实验不能覆盖所有VOC和NOx浓度范围,而数值模式则可以弥补烟雾箱实验的不足. 因此,在烟雾箱实验数据验证了MCM机制对苯和乙苯光氧化过程的基础上,通过MCM数值模拟得到一定环境条件下,苯和NOx在一定范围变化时的臭氧等值线,进而确定苯的臭氧生成活性和潜势是一个行之有效的方法. 首先用MCM模拟了烟雾箱实验条件下(即有效光强为0.238 min-1,温度300 K,相对湿度5%),苯和NOx的取值范围分别为0~4000×10-9 和 0~300×10-9时,苯-NOx光照体系中臭氧峰值等值线以及6 h时的臭氧等值线,结果如图 4(a)和4(c)所示(其中圆圈表示不同C6H6/NOx比值的实验点). 在考虑了MCM模拟值与实验值的偏差后,发现MCM模拟的臭氧等值区间与实验点能很好地吻合. 在此基础上,用实际太阳光照强度[J(NO2)=0.56 min-1], 在温度为300 K,相对湿度为50%条件下,对苯-NOx光氧化体系进行了进一步的模拟[图 4(b)和4(d)]. 发现当苯浓度在(10~50)×10-9,NOx浓度在(10~100)×10-9范围变化时,MCM模拟得到的臭氧峰值浓度为(4.2~65)×10-9. 扣除MCM模拟结果比实际高出20%(在其它苯光氧化实验中发现,相对湿度接近50%时,MCM模拟的臭氧浓度比实验值最多高出20%),则真实太阳光照下,苯在以上浓度区间内对臭氧峰值浓度(即苯的臭氧生成潜势)的贡献为:(3.5~54)×10-9. 在苯和NOx浓度相同的区间内,光照6 h后的臭氧值为(3.7~40)×10-9,扣除模拟值比实验值高出的20%, 6 h后臭氧的模拟值为(3.1~33)×10-9.
 | 图 4 MCM机制模拟实验室光强下以及自然光下苯光氧化产生的O3峰值等值线以及6 h O3等值线
Fig.4 MCM-predicted O3 peak and 6 h concentration isopleths from photo-oxidation of benzene under experimental and sunlight conditions
|
采用与苯相同的方法,通过MCM模拟得到了乙苯的臭氧等值线,进而确定其臭氧生成活性和潜势. 图 5(a)和5(c)为与苯相同的烟雾箱实验条件下,采用MCM机制模拟得到的乙苯光氧化体系的臭氧峰值以及6 h时臭氧浓度等值线,图中“○”为烟雾箱实验点. 可以看出实验点均落在等值线的脊线附近. 逐个比较了各个实验点上臭氧峰值浓度与MCM模拟值,发现除了90%相对湿度附近模拟值比实验值偏高以外,其它不同C8H10/NOx比率的数值模拟值均比实验值偏大10%~25%. 在比较了MCM模拟值与实验值的基础上,用与苯相同的模拟条件下得到乙苯在太阳光照条件下的臭氧峰值等值线和6 h时的臭氧等值线[图 5 (b)和5(d)]. 可以看出当乙苯在(10~50)×10-9、 NOx在(10~100)×10-9变化时,
 | 图 5 MCM机制模拟实验室光强下以及自然光下乙苯光氧化产生的臭氧峰值以及6 h浓度等值线
Fig.5 MCM-predicted O3 peak and 6 h concentration isopleths from photo-oxidation of ethylbenzene under experimental and sunlight conditions
|
MCM模拟得到的乙苯对臭氧最大值的贡献范围为:(4.6~198)×10-9; 对6 h时的臭氧贡献范围为(3.2~148)×10-9. 扣除MCM模拟值比实验值偏大的部分(按偏大21%计算),乙苯光氧化臭氧峰值浓度的贡献达(3.8~164)×10-9,在实际大气中光照6 h对臭氧的贡献达(2.6~122)×10-9.
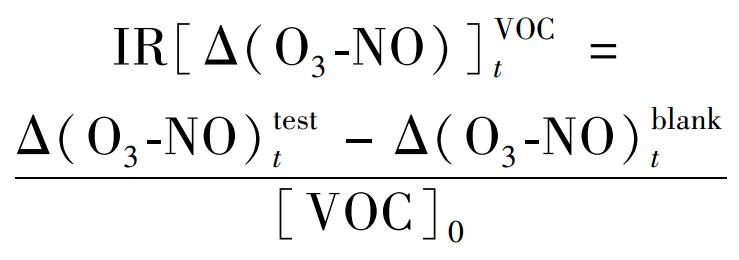
图 6为苯-NOx光氧化体系中NOx在(1~200)×10-9变化时得到的IR(O3 peak)max值和IR(t=6 h)max值随苯浓度变化的曲线. 为了节约计算时间,苯和NOx在(1~50)×10-9范围时,苯和NOx的增量为1×10-9; 当苯和NOx在(50~200)×10-9范围时,苯和NOx的增量为10×10-9. 结果表明,随着苯浓度从200×10-9到1×10-9不断减少,IR(t=6 h)max值不断增大,当苯初始浓度减小到9×10-9时,IR(t=6 h)max达到最大值1.47,即MIR值为1.47,平均每个碳对应的MIR值为0.25/C. Carter[15]用SAPRC机制计算得到苯在6 h时的MIR值为0.42/C,该值是本实验结果的1.7倍. 同时计算得到臭氧峰值时对应的IR(O3 peak)max值在苯初始浓度为10×10-9时出现了最大值4.4,即MOR为4.4,平均到每个碳以后MOR为0.73/C,而Carter[15]用SAPRC机制计算得到苯的MOR值为0.36/C. 通过MCM机制计算得到的MOR值是SAPRC机制的2倍. 这种差异是由于两种机制中关于苯的降解机制不同引起的. 由于MOR值反映了VOC降解全过程中对臭氧的最大贡献,因此其与碳键数量相关. 在MCM机制中考虑了由苯降解为CO2和H2 O的全过程. 而SAPRC 机制由于省略了许多中间降解过程,因此其可能低估了苯的MOR值. 通过苯的MIR值还可以计算苯在不同初始浓度时对臭氧的最大实际贡献量,即O3(t=6 h)Benzene=[Benzene]0×MIR. 在NOx初始浓度较高时,臭氧出现最大值的时间有时会超过12 h,因而MOR值反映的是苯的最大臭氧生成潜势.
采用与苯相同的方法,计算了乙苯的光氧化臭氧峰值增量反应活性MOR和6 h的增量反应活性IR(t=6 h)max,结果如图 6所示. 在乙苯和NOx分别从(1~200)×10-9变化时,计算得到乙苯MOR值为1.03/C,比Carter[15]用SAPRC机制计算的MOR值偏小37%. 在实验浓度范围内,当乙苯浓度为4×10-9时,IR(t=6 h)max达到峰值7.7,即平均每个碳原子后乙苯的MIR值为 0.97/C,而Carter[15]的MIR值为2.7/C,比本实验结果大了近3倍. 这说明采用SAPRC机制得到的MIR值可能高估了乙苯对臭氧的实际贡献.
 | 图 6 苯和乙苯氧化体系中6 h时IR极大值以及O3峰值时IR极大值随苯和乙苯浓度的变化 Fig.6 Changes of IR(t=6 h)max and IR(O3 peak)max in benzene and ethylbenzene systems |
比较苯和乙苯浓度的MIR值以及各自的MOR值可以看出,苯和乙苯光氧化体系中MIR值分别为0.25/C和0.97/C,乙苯对臭氧的实际贡献值是苯的3.9倍; MIR值反映了VOC物质的反应动力学活性,即其值的大小与其同OH自由基的反应快慢有关,即反应快的其MIR值较高. 在298 K时,乙苯和苯与OH自由基的反应速率常数分别为7.0×10-12 cm3 ·(molecules ·s)-1和1.2×10-12 cm3 ·(molecules ·s)-1,其比值约为5.8,而Carter[15]的MIR比值为6.3. 说明通过SAPRC机制得到的MIR值更接近苯和乙苯的反应动力学活性. 通过MCM计算得到的苯和乙苯对应的MOR值分别为0.73/C和1.03/C,乙苯的臭氧生成潜势是苯的1.4倍. MOR值反映的是VOC臭氧生成潜势. 根据文献[26]报道,在NO2存在时,VOC光氧化生成臭氧的潜势与C—C和C—H数目有关,即C—C键与C—H键数目越大则该物质最终完全氧化为CO2和H2 O时生成的O3越多. 苯与乙苯的C—C和C—H键总数分别为12和18,其比值为0.67. 根据MCM计算得到的苯和乙苯的MOR值之比为0.7,该比值与苯和乙苯的化学键数比值非常接近. 而相应的Carter[15]通过SAPRC机制得到MOR比值仅为0.22. 说明通过MCM得到的MOR值比由SAPRC得到的MOR值更能反映苯和乙苯的臭氧生成潜势. 此外,这还说明,与乙苯相比虽然苯氧化对臭氧的直接贡献很小,但是其主要氧化产物在进一步降解过程中对臭氧的贡献仍然很大.
(1)用烟雾箱研究了苯和乙苯在有NOx存在情况下的光氧化,并与MCM机制模拟结果进行了比较,发现MCM机制对干燥时苯和乙苯光氧化生成的臭氧模拟较好. 相对湿度≤5%时,在苯-NOx反应体系中,MCM模拟的臭氧最大值比实验值偏大20%,高湿时(83%)MCM模拟的臭氧峰值比实验值偏大41%. 在乙苯-NOx光氧化体系中,在相对湿度为5%~70%时,MCM模拟的臭氧峰值比实验值偏高约(10%~21%),湿度90%时MCM模拟值比实验偏大41%.
(2)在烟雾箱实验基础上,通过MCM机制模拟了太阳光照条件下,苯和乙苯在(0~200)×10-9,NOx在(0~200)×10-9范围内臭氧浓度等值线,扣除了MCM模拟偏差后,得到苯和乙苯的6 h臭氧贡献值分别为(3.1~33)×10-9和(2.6~122)×10-9,苯和乙苯的臭氧峰值范围分别是(3.5~54)×10-9和(3.8~164)×10-9.
(3)用MCM机制进一步模拟计算了苯和乙苯的MIR和MOR值,得到苯和乙苯的MIR分别为0.25/C和0.97/C. 得到苯和乙苯的MOR分别为0.73/C和1.03/C. 由于MCM机制考虑了苯系物降解的全部过程,因此用MCM得到的MOR值可能会更好地反映苯系物的臭氧潜势.
| [1] | Calvert J G, Atkinson R, Becker K H, et al. The mechanisms of atmospheric oxidation of aromatic hydrocarbons[M]. Oxford: Oxford University Press, 2002. |
| [2] | Watson J G, Chow J C, Fujita E M. Review of volatile organic compound source apportionment by chemical mass balance[J]. Atmospheric Environment, 2001, 35 (9): 1567-1584. |
| [3] | Yuan B, Shao M, Lu S, et al. Source profiles of volatile organic compounds associated with solvent use in Beijing, China[J]. Atmospheric Environment, 2010, 44 (15): 1919-1926. |
| [4] | 戴天有, 王玮, 任丽红, 等. 国产小汽车尾气非甲烷烃排放特征[J]. 中国科学(B辑): 化学, 2010, 40 (1): 76-85. |
| [5] | 张俊刚, 王跃思, 王珊, 等. 京津地区大气中非甲烷烃(NMHCs)质量浓度水平和反应活性研究[J]. 环境科学研究, 2008, 21 (5): 158-162. |
| [6] | Goldan P D, Kuster W C, Fehsenfeld F C, et al. Hydrocarbon measurements in the southeast United States: the rural oxidants in the southern environment program 1990[J]. Journal of Geophysical Research, 1995, 100 (D12): 25945-25963. |
| [7] | Jang M, Kamens R M. Characterization of secondary aerosol from the photooxidation of toluene in the presence of NOx and 1-propene[J]. Environmental Science & Technology, 2001, 35 (18): 3626-3639. |
| [8] | 吴方堃, 王跃思, 安俊琳, 等. 北京奥运时段VOCs浓度变化、臭氧产生潜势及来源分析研究[J]. 环境科学, 2010, 31 (1): 10-16. |
| [9] | 陆思华, 白郁华, 陈运宽, 等. 北京市机动车排放挥发性有机化合物的特征[J]. 中国环境科学, 2003, 23 (2): 127-130. |
| [10] | Duan J, Tan J, Yang L, et al. Concentration, sources and ozone formation potential of volatile organic compounds (VOCs) during ozone episode in Beijing[J]. Atmospheric Research, 2008, 88 (1): 25-35. |
| [11] | Zou S C, Lee S C, Chan C Y, et al. Characterization of ambient volatile organic compounds at a landfill site in Guangzhou, South China[J]. Chemosphere, 2003, 51 (9): 1015-1022. |
| [12] | 陈洪伟, 李攻科, 李核, 等. 广州地区大气中挥发性有机物的污染状况[J]. 环境化学, 2003, 22 (1): 89-92. |
| [13] | Derwent R G, Jenkin M E, Saunders S M. Photochemical ozone creation potentials for a large number of reactive hydrocarbons under European conditions[J]. Atmospheric Environment, 1996, 30 (2): 181-199. |
| [14] | Derwent R G, Jenkin M E, Saunders S M, et al. Photochemical ozone creation potentials for organic compounds in northwest Europe calculated with a Master Chemical Mechanism[J]. Atmospheric Environment, 1998, 32 (14-15): 2429-2441. |
| [15] | Carter W P L. Development of ozone reactivity scales for volatile organic compounds[J]. Journal of the Air & Waste Management Association, 1994, 44 (7): 881-899. |
| [16] | Carter W P L. Computer modeling of environmental chamber measurements of maximum incremental reactivities of volatile organic compounds[J]. Atmospheric Environment, 1995, 29 (18): 2513-2527. |
| [17] | Jenkin M E, Saunders S M, Wagner V, et al. Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part B): tropospheric degradation of aromatic volatile organic compounds[J]. Atmospheric Chemistry and Physics, 2003, 3: 181-193. |
| [18] | Kelly N A, Chang T Y. An experimental investigation of incremental reactivities of volatile organic compounds[J]. Atmospheric Environment, 1999, 33 (13): 2101-2110. |
| [19] | Carter W P L. Documentation for the SAPRC atmospheric photochemical mechanism preparation and emissions processing programs for implementation in airshed models[R]. 1988: 214. |
| [20] | Saunders S M, Jenkin M E, Derwent R G, et al. Protocol for the development of the Master Chemical Mechanism, MCMv3 (PartA): tropospheric degradation of non-aromatic volatile organic compounds[J]. Atmospheric Chemistry and Physics, 2003, 3: 161-180. |
| [21] | 石玉珍, 徐永福, 贾龙. 大气化学机理的发展及应用[J]. 气候与环境研究, 2012, 17 (1): 112-124. |
| [22] | 贾龙, 徐永福, 石玉珍. 光化学烟雾箱的表征及初步应用[J]. 环境科学, 2011, 32 (1): 351-361. |
| [23] | 黄丽华, 莫创荣, 徐永福, 等. 大气中丙烷光氧化臭氧生成活性的烟雾箱模拟[J]. 环境科学, 2012, 33 (8): 2551-2557. |
| [24] | Jia L, Xu Y F, Shi Y Z. Investigation of the ozone formation potential for ethanol using a smog chamber[J]. China Science Bulletin, 2012, 57 (34): 4472-4481. |
| [25] | Carter W P L, Pierce J A, Luo D M, et al. Environmental chamber study of maximum incremental reactivities of volatile organic compounds[J]. Atmospheric Environment, 1995, 29 (18): 2499-2511. |
| [26] | Jenkina M E, Watson L A, Utembe S R, et al. A Common Representative Intermediates (CRI) mechanism for VOC degradation. Part 1: Gas phase mechanism development[J]. Atmospheric Environment, 2008, 42 (31): 7185-7195. |